Advanced Usage
This section describes the various features and options of ClipKIT.
Modes
This section describes the trimming modes implemented in ClipKIT. If you are unsure which is appropriate for you, we recommend using the default smart-gap trimming mode.
ClipKIT can be run with fifteen different modes, which are specified with the -m/–mode argument. Default: ‘smart-gap’ (or ‘gappy’ when -g/–gaps is provided without -m)
smart-gap: dynamic determination of gaps threshold
entropy: trim sites above a normalized Shannon entropy threshold (default: 0.8)
gappy: trim all sites that are above a threshold of gappyness (default: 0.9)
block-gappy: trim contiguous runs of sites above a threshold of gappyness (default: 0.9)
gappyout: infer a gap threshold from alignment-wide gap distribution and trim above it (gappyout-inspired behavior; not strict trimAl compatibility)
composition-bias: trim sites above a normalized composition-bias threshold (default: 0.8)
heterotachy: infer a parsimony guide tree and trim sites with high clade-to-clade entropy variation (default: 0.8)
kpic: keep only parsimony informative and constant sites
kpic-smart-gap: a combination of kpic- and smart-gap-based trimming
kpic-gappy: a combination of kpic- and gappy-based trimming
kpi: keep only parsimony informative sites
kpi-smart-gap: a combination of kpi- and smart-gap-based trimming
kpi-gappy: a combination of kpi- and gappy-based trimming
c3: remove third codon position from alignment
cst: custom site trimming (remove sites specified by the user)
# smart-gap-based trimming
clipkit <input>
clipkit -m smart-gap
# gappy-based trimming
clipkit <input> -m gappy
# block-gappy trimming
clipkit <input> -m block-gappy
# gappyout-style trimming
clipkit <input> -m gappyout
# entropy-based trimming
clipkit <input> -m entropy
# composition-bias trimming
clipkit <input> -m composition-bias
# heterotachy trimming (parsimony guide tree based)
clipkit <input> -m heterotachy
# kpic-based trimming
clipkit <input> -m kpic
# kpic- and smart-gap-based trimming
clipkit <input> -m kpic-smart-gap
# kpic- and gappy-based trimming
clipkit <input> -m kpic-gappy
# kpi-based trimming
clipkit <input> -m kpi
# kpi- and smart-gap-based trimming
clipkit <input> -m kpi-smart-gap
# kpi- and gappy-based trimming
clipkit <input> -m kpi-gappy
# remove third codon position
clipkit <input> -m c3
# conduct site-specific trimming
clipkit <input> -m cst -a <auxiliary file>
Output
By default, output files will have the same name as the input file with the suffix “.clipkit” appended to the name. Users can specify output file names with the -o option.
# specify output
clipkit <input> -o <output>
Log
It can be useful to have information about each position in an alignment. For example, this information could be used in alignment diagnostics, fine-tuning of trimming parameters, etc. To create the log file, use the -l/\-\-log option. Using this option will create a four-column file with the suffix ‘clipkit.log’. Default: off
col1: position in the alignment (starting at 1)
col2: reports if site was trimmed or kept (trim or keep, respectively)
col3: reports if the site is parsimony informative or not (PI or nPI, respectively)
col4: reports the effective unavailable fraction of the position. In the default
missingambiguity mode this includes configured gaps and recognized IUPAC ambiguity symbols; otherwise it includes configured gaps.
clipkit <input> -l
Complementary
Having an alignment of the sequences that were trimmed can be useful for other analyses. To obtain an alignment of the sequences that were trimmed, use the -c/\-\-complementary option.
clipkit <input> -c
Output file with the suffix ‘.clipkit.complement’
Codon
Trims codon-based alignments. If one position in a codon should be trimmed, the whole codon will be trimmed. To conduct codon-based trimming, use the -co/\-\-codon argument.
clipkit <input> --codon
# or
clipkit <input> -co
Stop codon masking
For codon-aligned nucleotide MSAs, ClipKIT can replace selected in-frame stop
codons with --- before calculating gap statistics or choosing columns to
trim. Masking preserves sequence lengths and alignment rectangularity. DNA
stops (TAA, TAG, and TGA) and their RNA equivalents are recognized
case-insensitively.
The --remove_stop_codons modes are:
terminal: mask a stop only when it is the final complete, non-gap codon in a sequence. Trailing gap codons are allowed.internal: mask all in-frame stops other than a terminal stop.all: mask both terminal and internal stops.
This option requires --codon and nucleotide input. The alignment length
must be divisible by three. Gapped or incomplete codons are not interpreted as
stops. Because masking occurs first, the new gaps participate in gap-based
trimming; when a masked codon column reaches the selected gap threshold,
codon-aware trimming can remove that column from every sequence.
# mask terminal stops only
clipkit coding.fa --codon --sequence_type nt --remove_stop_codons terminal
# mask internal stops only
clipkit coding.fa --codon --sequence_type nt --remove_stop_codons internal
# mask terminal and internal stops, then apply a gap threshold
clipkit coding.fa --codon --sequence_type nt --remove_stop_codons all -m gappy -g 0.5
Execution output reports separate terminal and internal masking counts. JSON
reports created with --report_json contain the selected mode and the same
counts under stop_codon_masking. The Python API accepts terminal,
internal, or all through its remove_stop_codons argument.
from clipkit import clipkit
trim_run, stats = clipkit(
input_file_path="coding.fa",
mode="gappy",
gaps=0.9,
sequence_type="nt",
codon=True,
remove_stop_codons="all",
)
print(trim_run.stop_codon_masking.summary)
Custom site trimming (cst mode)
Custom site trimming specified using a tab-delimited text file specified using the -a argument.
clipkit <input> -m cst -a <auxiliary_file>
The auxiliary_file is a two column tab-delimited file wherein the first column is the site (starting at 1) and the second column specifies if the site should be kept or trimmed using the strings “keep” or “trim”.
cat auxiliary_file.txt
1 keep
2 trim
3 keep
4 keep
5 keep
6 keep
Alternatively, users can specify sites that are only kept or trimmed using the auxiliary_file. For example, the following would be equivalent to the auxiliary file described above.
cat auxiliary_file.txt
2 trim
Similarly, the following would conduct the trimming, wherein the second site is removed but all others are kept.
cat auxiliary_file.txt
1 keep
3 keep
4 keep
5 keep
6 keep
Gaps
Positions with gappyness greater than threshold will be trimmed.
Must be between 0 and 1. (Default: 0.9). When -g is provided without
-m, the trimming mode automatically switches from smart-gap to gappy
so that the threshold is honoured. If -m is explicitly set to a mode that
dynamically determines its own threshold (smart-gap, kpi-smart-gap,
kpic-smart-gap, or gappyout), the -g value is ignored and a warning
is printed. This argument is also ignored when using the kpi and kpic modes.
In entropy mode, this value is treated as a normalized Shannon entropy threshold
(default: 0.8).
With the default --ambiguity_handling missing policy, gappyness is an
effective unavailable fraction: the union of configured gap characters and
recognized IUPAC ambiguity symbols divided by the number of sequences. The
configured-gap and ambiguity fractions remain separately visible in HTML trim
reports. A symbol can occur in both tracks when it is both a recognized
ambiguity code and a configured gap character.
To specify a gaps threshold, use the -g/\-\-gaps argument.
clipkit <input> --gaps 0.4
# or
clipkit <input> -g 0.4
Gap Characters
Specifies gap characters used in the input file. For example, “NnXx-?” would specify that “N”, “n”, “X”, “x”, “-”, and “?” are gap characters. Note, the first gap character cannot be “-” because the parser will interpret the gaps list as a new argument.
clipkit <input> -gc NnXx-?
Sequence Type
Specifies the type of sequences in the input file. The default is auto-detection of sequence type. Valid options include aa or nt for amino acids and nucleotides. This argument is case insensitive. This matters for what characters are considered gaps. For amino acids, -, ?, *, and X are considered gaps. For nucleotide sequences, the same characters are considered gaps as well as N.
clipkit <input> -s aa
Use this option to specify that input sequences are amino acids.
clipkit <input> -s nt
Use this option to specify that input sequences are nucleotides.
Ambiguity handling
ClipKIT recognizes the IUPAC nucleotide ambiguity symbols R, Y,
S, W, K, M, B, D, H, V, N, and X.
For proteins it recognizes B (D or N), Z (E or Q),
J (I or L), and X (any standard amino acid). Recognition is
case-insensitive. IUPAC-rich nucleotide alignments are also recognized by
automatic sequence-type detection.
Use --ambiguity_handling (or --ambiguity-handling) to select one of
three policies:
missing(default): ambiguity symbols do not contribute states to entropy, composition-bias, heterotachy entropy, or KPI/KPIC classification. They do contribute to the effective unavailable fraction used by gappy, block-gappy, gappyout, smart-gap, and combined gap/classification modes. Sites with no analyzable states are removed by entropy, composition-bias, and heterotachy modes.fractional: an ambiguity symbol contributes equal weight to each possible state for entropy, composition-bias, and heterotachy entropy. For example, nucleotideRcontributes 0.5 toAand 0.5 toG. Ambiguity symbols are still excluded from KPI/KPIC site classification, so uncertainty alone cannot create a parsimony-informative site. Gap-based modes count configured gap characters only.literal: each non-gap ambiguity symbol is treated as its own state. This reproduces ClipKIT’s legacy ambiguity interpretation. Gap-based modes count configured gap characters only.
Configured gap characters always take precedence over the selected ambiguity
policy. Consequently, N and X remain gaps under the default nucleotide
gap-character set, and X remains a gap under the default protein set. To
fractionally expand those symbols, provide a gap-character set that does not
contain them (for example, -gc '?*-').
# conservative default
clipkit nucleotide.fa --sequence_type nt --ambiguity_handling missing
# fractionally weight partial ambiguity codes
clipkit nucleotide.fa --sequence_type nt --ambiguity_handling fractional
# reproduce the historical literal-state behavior
clipkit nucleotide.fa --sequence_type nt --ambiguity_handling literal
These policies affect analysis only. ClipKIT never rewrites ambiguity symbols
in the kept or complementary alignment output. JSON reports, HTML trim reports,
and the Python TrimRun object record the selected policy. HTML reports also
provide per-site configured-gap, ambiguity, and resolved-state fractions.
Ends only
For a given trimming mode, this option trims only sites at the ends of an alignment. For example, if the sites that should be trimmed include [0, 1, 2, 4, 5, 6, 14, 15, 16] for smart-gap mode and an alignment of length 16, adding the ends_only mode will result in [0, 1, 2, 14, 15, 16] being the sites that will be trimmed. Use this argument with -eo, --ends_only.
clipkit <input> -eo
# or
clipkit <input> --ends_only
Threads
ClipKIT supports parallel processing for site classification and character frequency calculations. For larger alignments, this can significantly speed up processing.
The number of threads can be specified using the -t/\-\-threads argument. Default: 1
# Single-threaded processing (default)
clipkit <input>
# Multi-threaded processing with 4 threads
clipkit <input> -t 4
# or
clipkit <input> --threads 4
Performance Notes:
Parallel processing is activated adaptively based on alignment size and requested threads
For smaller alignments, single-threaded mode is typically faster due to multiprocessing overhead
The optimal number of threads depends on your system and alignment size
For KPI/KPIC family modes (kpi, kpi-gappy, kpi-smart-gap, kpic, kpic-gappy, kpic-smart-gap), ClipKIT may automatically use fewer threads than requested when that is expected to be faster
Results are identical regardless of the number of threads used (fully reproducible)
Dry run
Use dry run mode to execute trimming and compute summary statistics without writing alignment, complementary, or log output files.
clipkit <input> --dry_run
Validate only
Use validate-only mode to check input format and argument consistency (including
auxiliary file checks for cst mode) and then exit without trimming.
clipkit <input> --validate_only
Report JSON
Write a machine-readable JSON report with run configuration and outcome details.
# explicit report path
clipkit <input> --report_json run_report.json
# default report path: <output>.report.json
clipkit <input> --report_json
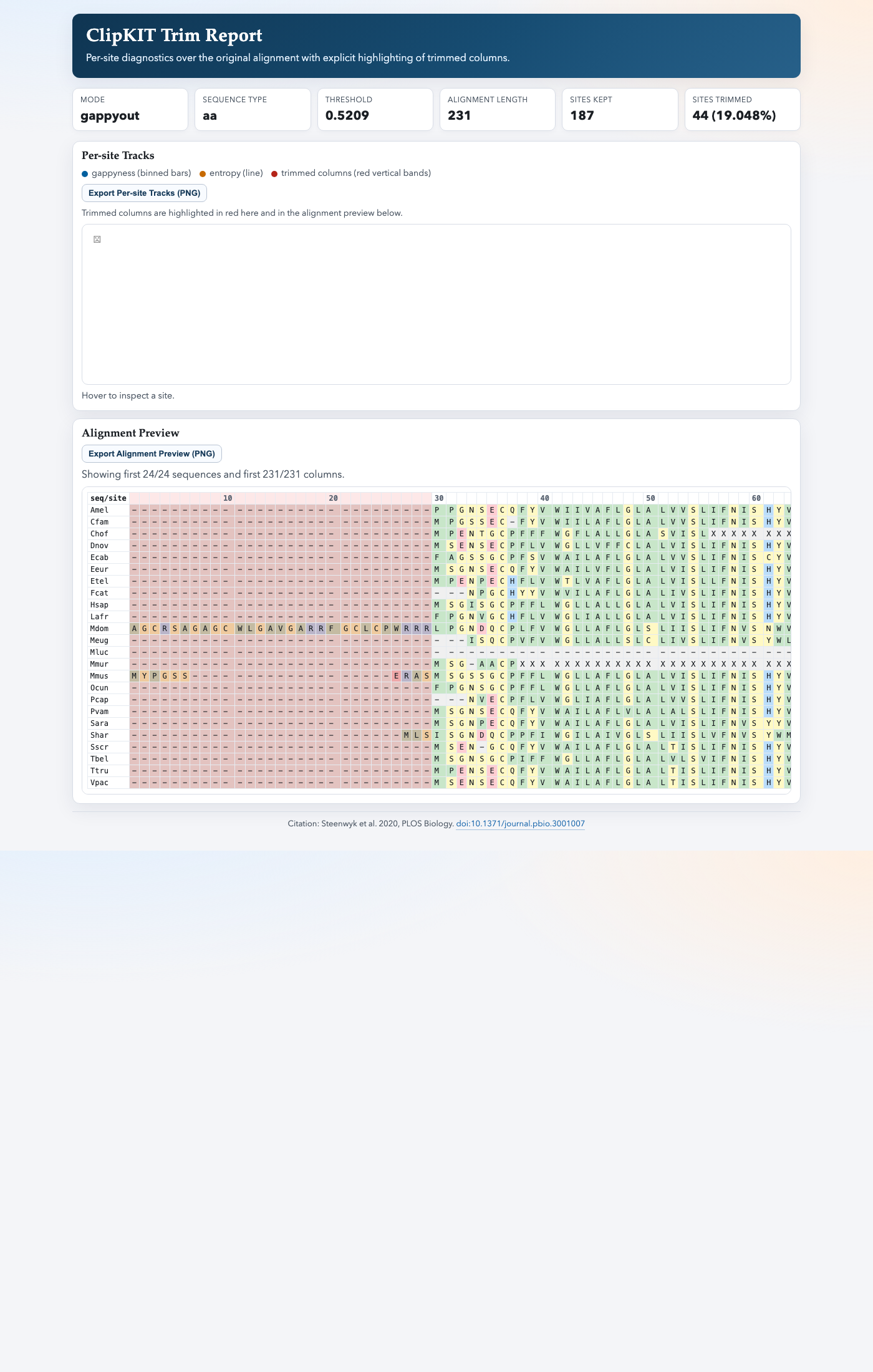
Plot trim report
Write an interactive HTML report with per-site tracks and trimmed-column highlighting.
# explicit report path
clipkit <input> --plot_trim_report run_plot.html
# default report path: <output>.trim_report.html
clipkit <input> --plot_trim_report
The report includes:
Per-site effective-unavailable bars and an entropy line plot
Per-site configured-gap, ambiguity, and resolved-state fractions in the embedded report data and hover diagnostics
Highlighting of trimmed columns in both tracks and alignment preview
Amino-acid or nucleotide coloring in the alignment preview (auto-detected)
Export buttons for saving per-site tracks and alignment preview as PNG files
Example preview:
All options
Option |
Usage and meaning |
|---|---|
|
Print help message. |
|
Print software version. |
|
Specify trimming mode (including |
|
Specify output file name. |
|
Specify threshold (between 0 and 1): gappyness for most modes, normalized entropy for |
|
Specify gap characters used in input file (AAs: |
|
Conduct codon-based trimming. Default: off. |
|
Mask selected in-frame stop codons as gaps before trimming. Requires nucleotide input and |
|
Specify sequence type of input file ( |
|
Control how recognized IUPAC ambiguity symbols contribute to analysis. Default: missing. |
|
Specify input file format*. Default: auto-detect. |
|
Specify output file format*. Default: input file type. |
|
Create a log file. Default: off. |
|
Create a complementary alignment file. Default: off. |
|
Auxiliary file used for specifying sites to trim in |
|
Trim only sites at alignment ends that would otherwise be removed. Default: off. |
|
Disable logging to stdout. Default: off. |
|
Requested threads for parallel processing; KPI/KPIC modes may auto-tune lower. Default: 1. |
|
Run trimming/stat calculations but skip writing output files. Default: off. |
|
Validate inputs/arguments and exit without trimming. Default: off. |
|
Write a JSON run report; if no path is given, uses |
*Acceptable file formats include: fasta, clustal, maf, mauve, phylip, phylip-sequential, phylip-relaxed, stockholm