Faidx
Extract entries from FASTA files
Command identity
- Canonical command:
faidx- Handler:
faidx- Aliases:
ge, get_entry
- Standalone executables:
pk_faidx, pk_ge, pk_get_entry
- Categories:
Alignment & dataset utilities
Runtime interface
Synopsis
phykit faidx <fasta> [--entry <entry>] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Unless the guidance below states otherwise, results are emitted as command output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
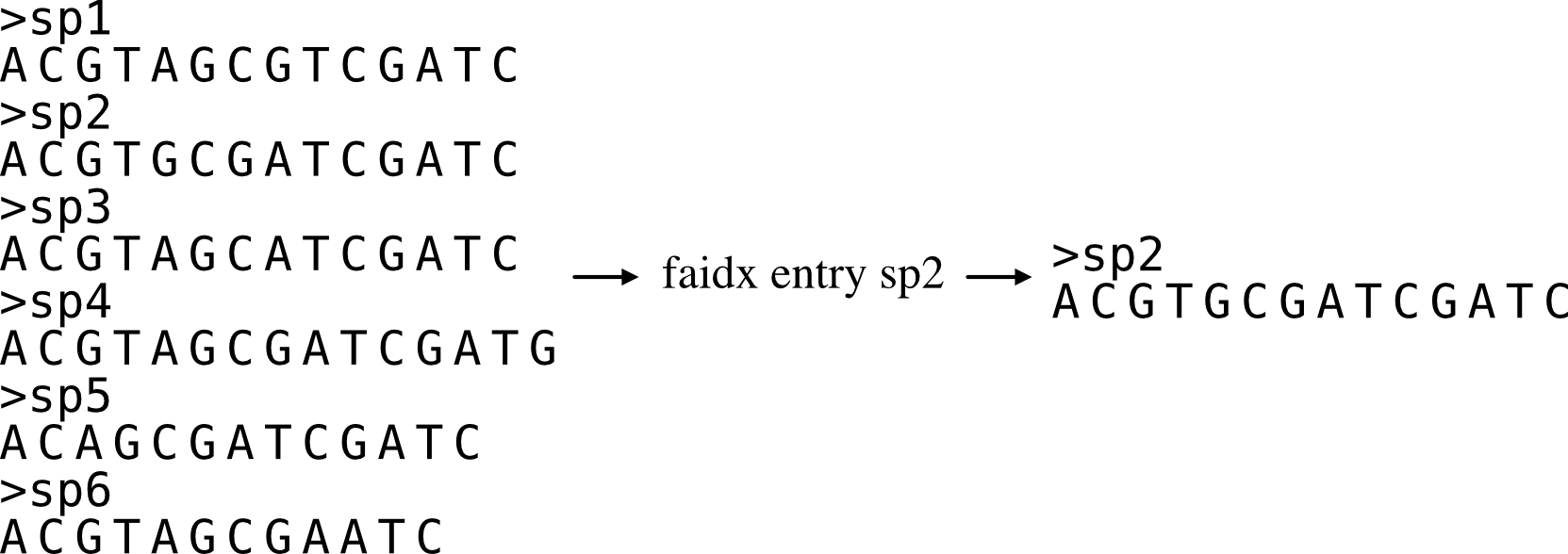
Extracts a sequence entry from a fasta file.
This function works similarly to the faidx function in samtools, but does not require an indexing step.
To obtain multiple entries, input multiple entries separated by a comma (,). For example, if you want entries named "seq_0" and "seq_1", the string "seq_0,seq_1" should be associated with the -e argument.
phykit faidx <fasta> -e/--entry <fasta entry> [--json]
Options:
<fasta>: first argument after function name should be a fasta file
-e/--entry: entry name to be extracted from the inputted fasta file
--json: optional argument to print results as JSON