Phylogenetic signal
Test for phylogenetic signal in traits (supports discordance-aware VCV with -g)
Command identity
- Canonical command:
phylogenetic_signal- Handler:
phylogenetic_signal- Aliases:
phylo_signal, ps
- Standalone executables:
pk_phylogenetic_signal, pk_phylo_signal, pk_ps
- Categories:
Phylogenetic signal
Runtime interface
Synopsis
phykit phylogenetic_signal --tree <tree> --trait_data <trait_data> [--method <method>] [--permutations <permutations>] [--gene-trees <gene_trees>] [--multivariate] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
false |
str |
blombergs_k |
blombergs_k, lambda |
|
false |
int |
1000 |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Unless the guidance below states otherwise, results are emitted as command output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
Calculate phylogenetic signal for continuous trait data on a phylogeny.
Two methods are available:
Blomberg's K (Blomberg et al. 2003): measures the degree of phylogenetic signal relative to expectation under Brownian motion. K = 1 indicates trait variation consistent with BM; K < 1 indicates less phylogenetic signal than expected; K > 1 indicates more. P-value is computed via permutation test.
Pagel's lambda (Pagel 1999): a tree-scaling parameter estimated by maximum likelihood. Lambda = 0 indicates no phylogenetic signal; lambda = 1 indicates trait evolution consistent with BM. P-value is computed via likelihood ratio test against lambda = 0.
The trait file should be tab-delimited with two columns (taxon_name<tab>trait_value). Lines starting with '#' are treated as comments. If the tree and trait file have different taxa, the intersection is used and warnings are printed to stderr.
Multivariate K_mult (Adams 2014): When the --multivariate flag is used,
K_mult is computed instead of single-trait K. This generalizes Blomberg's K to
multivariate data using distances in trait space. The trait file should be a
multi-column TSV with a header row (taxon<tab>trait1<tab>trait2<tab>...),
the same format used by phylogenetic_ordination and trait_correlation.
K_mult only works with Blomberg's K framework; combining --multivariate
with --method lambda will produce an error.
Output for Blomberg's K: K_value<tab>p_value<tab>R2_phylo
Output for Pagel's lambda: lambda_value<tab>log_likelihood<tab>p_value<tab>R2_phylo
Output for K_mult: K_mult<tab>p_value<tab>n_traits<tab>permutations
R²_phylo compares the fitted trait variance under Brownian motion with the
variance under a white-noise model: R²_phylo = 1 - (σ²_BM / σ²_WN).
Values near 1 indicate a large relative reduction in fitted variance under the
Brownian-motion model. This statistic is not a literal percentage of trait
variance caused by phylogenetic relatedness and is not Pagel's lambda.
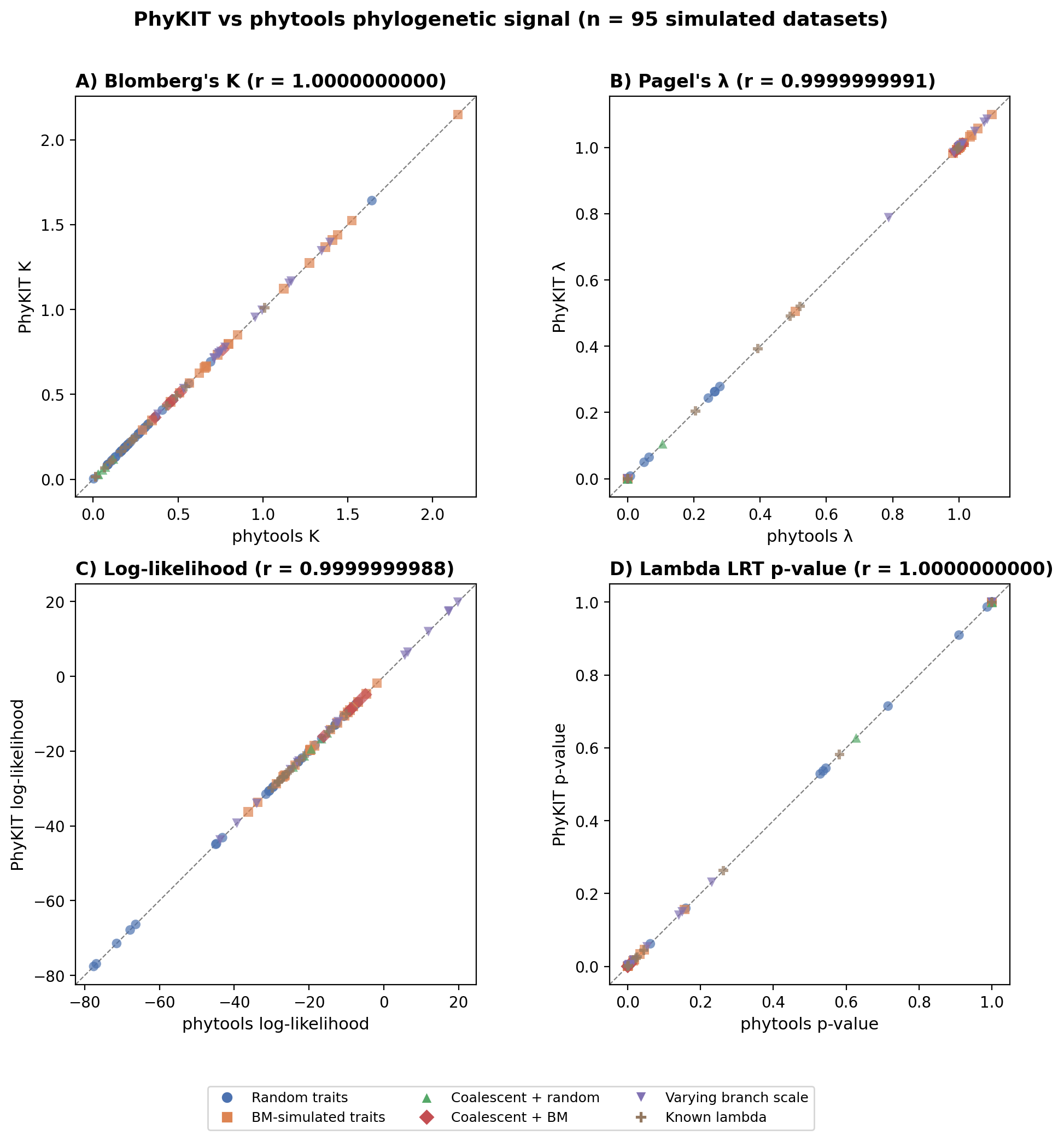
Results have been validated against the R package phytools (phylosig function)
across 95 simulated datasets spanning diverse tree sizes (5-50 tips), topologies
(pure-birth, coalescent), trait models (random, Brownian motion, known lambda),
and branch length scales. All metrics show Pearson r > 0.999 with phytools.
phykit phylogenetic_signal -t <tree> -d <trait_data> [-m <method>] [-p <permutations>] [-g <gene_trees>] [--multivariate] [--json]
Options:
-t/--tree: a tree file in Newick format
-d/--trait_data: tab-delimited trait file (taxon_name<tab>trait_value)
-m/--method: method to use: blombergs_k or lambda (default: blombergs_k)
-p/--permutations: number of permutations for Blomberg's K (default: 1000)
-g/--gene-trees: optional multi-Newick file of gene trees; when provided, uses a discordance-aware VCV (genome-wide average) instead of the species-tree VCV
--multivariate: compute K_mult (Adams 2014) for multivariate traits; the trait file should be a multi-column TSV with header row instead of two-column
--json: optional argument to print results as JSON
R validation: Validated against phytools, geiger in R
(see tests/r_validation/validate_signal_r2.R and tests/r_validation/validate_kmult.R).