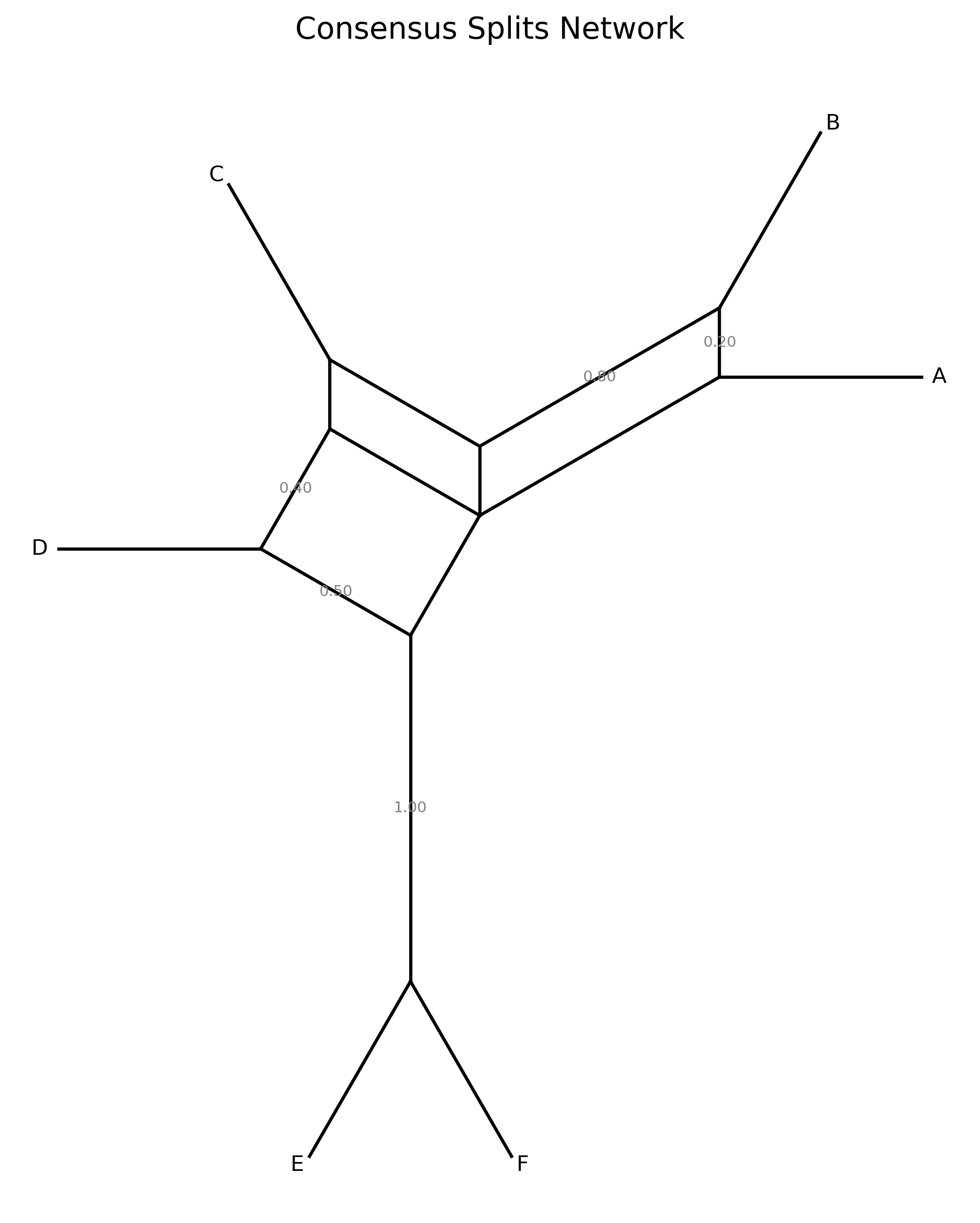
Consensus network
Consensus network from multiple trees
Command identity
- Canonical command:
consensus_network- Handler:
consensus_network- Aliases:
consnet, splitnet, splits_network
- Standalone executables:
pk_consensus_network, pk_consnet, pk_splitnet, pk_splits_network
- Categories:
Tree comparison & consensus
Runtime interface
Synopsis
phykit consensus_network --trees <trees> [--threshold <threshold>] [--missing-taxa <missing_taxa>] [--plot-output <plot_output>] [--max-splits <max_splits>] [--histogram <histogram>] [--fig-width <fig_width>] [--fig-height <fig_height>] [--dpi <dpi>] [--no-title] [--title <title>] [--legend-position <legend_position>] [--ylabel-fontsize <ylabel_fontsize>] [--xlabel-fontsize <xlabel_fontsize>] [--title-fontsize <title_fontsize>] [--axis-fontsize <axis_fontsize>] [--colors <colors>] [--ladderize] [--cladogram] [--circular] [--color-file <color_file>] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
false |
float |
0.1 |
any |
|
false |
str |
allow |
allow, error, shared |
|
false |
str |
none |
any |
|
false |
int |
30 |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
int |
300 |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Output-file options: --plot-output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
Extract bipartition splits from a collection of gene trees and summarize conflicting phylogenetic signal. Counts how frequently each non-trivial bipartition appears across input trees and filters by a minimum frequency threshold. Optionally draws a circular splits network diagram.
Polytomies (collapsed branches) in input trees are handled conservatively: splits from polytomous nodes are excluded since they represent unresolved relationships. Trifurcating roots (standard unrooted Newick) are not affected. This allows gene trees with collapsed low-support branches to be used directly as input.
Input can be either: 1) a file with one Newick tree per line, or 2) a file with one tree-file path per line.
phykit consensus_network -t/--trees <trees> [--threshold 0.1] [--missing-taxa allow|error|shared] [--plot-output <file>]
[--max-splits <int>] [--histogram <file>]
[--fig-width <float>] [--fig-height <float>] [--dpi <int>] [--no-title] [--title <str>]
[--legend-position <str>] [--ylabel-fontsize <float>] [--xlabel-fontsize <float>]
[--title-fontsize <float>] [--axis-fontsize <float>] [--colors <str>] [--ladderize] [--cladogram] [--circular] [--color-file <file>] [--json]
Options:
-t/--trees: file containing trees (one Newick per line) or tree-file paths (one per line)
--threshold: minimum split frequency to include, between 0 and 1 (default: 0.1)
--missing-taxa: handling strategy for mismatched taxa: allow keeps each
tree's available taxa, shared prunes to the intersection, and error
rejects mismatched sets (default: allow)
--plot-output: output filename for the circular splits network plot (optional)
--max-splits: maximum number of splits drawn in the network graph; all
qualifying splits remain in text and JSON output (default: 30)
--histogram: optional output filename for a split-frequency histogram
--fig-width: figure width in inches (auto-scaled if omitted)
--fig-height: figure height in inches (auto-scaled if omitted)
--dpi: resolution in DPI (default: 300)
--no-title: hide the plot title
--title: custom title text
--legend-position: legend location (e.g., "upper right", "none" to hide)
--ylabel-fontsize: font size for y-axis labels; 0 to hide
--xlabel-fontsize: font size for x-axis labels; 0 to hide
--title-fontsize: font size for the title
--axis-fontsize: font size for axis labels
--colors: comma-separated colors (hex or named)
--ladderize: ladderize (sort) the tree before plotting
--cladogram: draw cladogram (equal branch lengths, tips aligned) instead of phylogram
--circular: draw circular (radial/fan) phylogram instead of rectangular
--color-file: color annotation file for tip labels, clade ranges, and branch colors (iTOL-inspired TSV format)
--json: optional argument to print results as JSON
When --plot-output is specified, a circular splits network diagram is produced.
Taxa are arranged at equal angles around a circle. Each split is drawn as a chord
connecting the boundary points between the two sides of the bipartition. Chord
thickness and opacity scale with split frequency — thicker, darker lines indicate
splits supported by more gene trees.