Phylogenetic path analysis
Compare causal DAGs via d-separation + PGLS (von Hardenberg & Gonzalez-Voyer 2013)
Command identity
- Canonical command:
phylo_path- Handler:
phylo_path- Aliases:
phylopath, ppath
- Standalone executables:
pk_phylo_path, pk_phylopath, pk_ppath
- Categories:
Phylogenetic comparative methods
Runtime interface
Synopsis
phykit phylo_path --tree <tree> --traits <traits> --models <models> [--best-only] [--plot-output <plot_output>] [--csv <csv>] [--fig-width <fig_width>] [--fig-height <fig_height>] [--dpi <dpi>] [--no-title] [--title <title>] [--legend-position <legend_position>] [--ylabel-fontsize <ylabel_fontsize>] [--xlabel-fontsize <xlabel_fontsize>] [--title-fontsize <title_fontsize>] [--axis-fontsize <axis_fontsize>] [--colors <colors>] [--ladderize] [--cladogram] [--circular] [--color-file <color_file>] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
int |
300 |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Output-file options: --plot-output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
Phylogenetic path analysis (von Hardenberg & Gonzalez-Voyer 2013). Compare competing causal hypotheses (DAGs) about trait evolution while accounting for phylogenetic non-independence, using Shipley's d-separation test combined with PGLS.
How it works:
Define a set of candidate causal models as directed acyclic graphs (DAGs). Each DAG specifies which traits causally influence which other traits.
For each DAG, derive the basis set — the minimal set of conditional independence claims implied by the graph (Shipley 2000).
Test each independence claim using PGLS with Pagel's lambda. If the predictor is non-significant after conditioning, the independence holds.
Combine p-values into Fisher's C statistic. A model with a high p-value (C-test) is consistent with the data; a low p-value means the model is rejected.
Rank models by CICc (C-statistic Information Criterion, corrected for sample size). Lower CICc = better fit.
Estimate path coefficients (standardized PGLS regression slopes) and conditionally average them across well-supported models weighted by CICc.
Interpreting the output:
CICc and weights: models with delta CICc < 2 have substantial support. Weights sum to 1 and represent relative evidence.
Model p-value: p > 0.05 means the data are consistent with the DAG's independence claims. A rejected model (p < 0.05) has causal structure contradicted by the data.
Path coefficients: standardized (z-scored) regression slopes. Positive = positive causal effect; magnitude indicates strength. CIs not overlapping zero indicate significance.
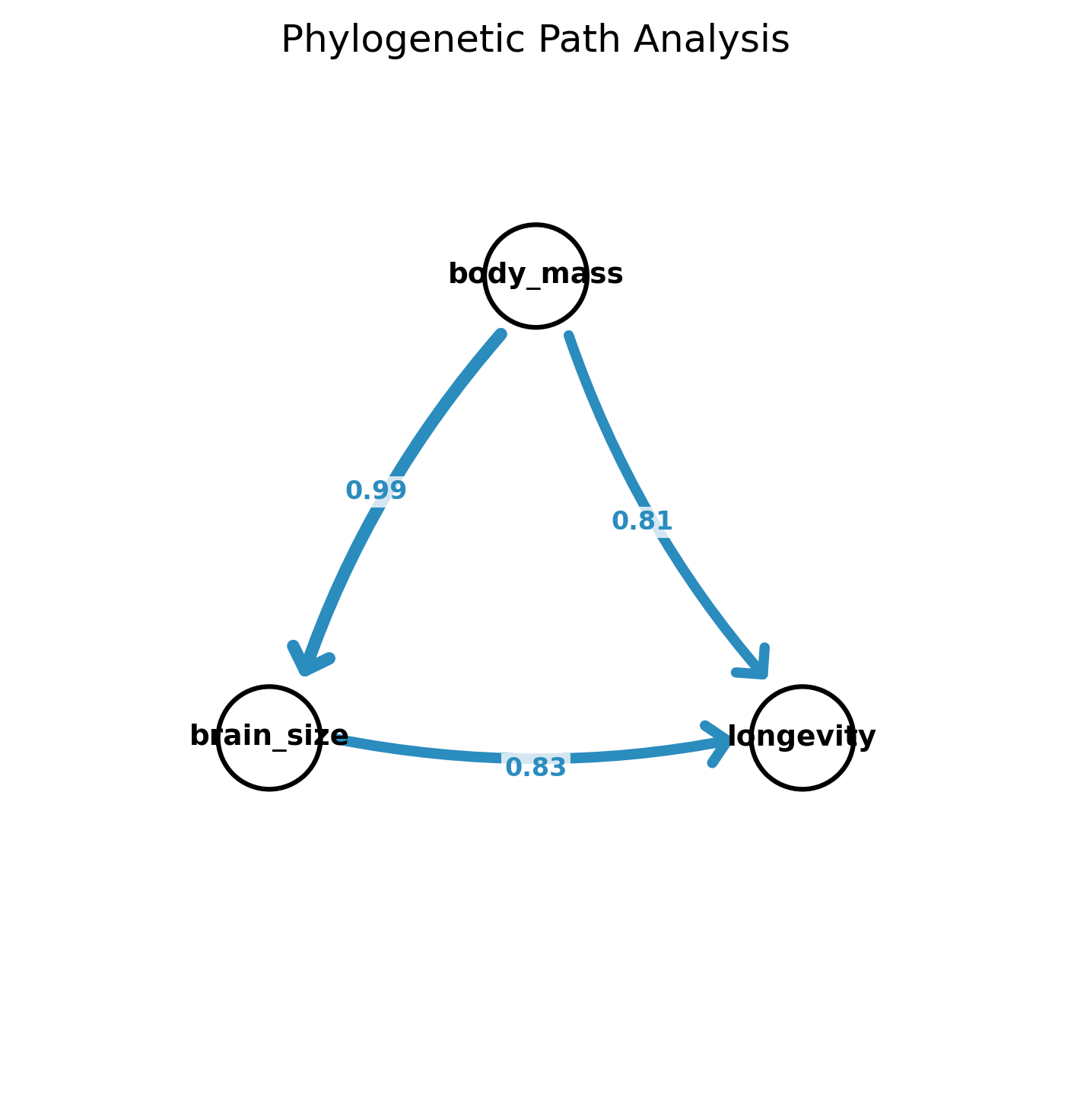
DAG plot showing model-averaged path coefficients. Blue = positive effect, line thickness proportional to coefficient magnitude.
Model file format:
# model_name: edge1, edge2, ...
direct: body_mass->brain_size, body_mass->longevity
mediated: body_mass->brain_size, brain_size->longevity
full: body_mass->brain_size, body_mass->longevity, brain_size->longevity
phykit phylo_path -t <tree> --traits <traits_file> --models <models_file>
[--best-only] [--plot-output <file>] [--csv <file>] [--json]
Options:
-t/--tree: species tree file (required)
--traits: TSV file with taxon and continuous trait columns (required)
--models: model definition file with candidate DAGs (required)
--best-only: report only best model coefficients instead of model averaging
--plot-output: output DAG plot file (.png, .pdf, .svg)
--csv: output CSV with model comparison table and path coefficients
--json: output results as JSON
R validation: Fisher's C, CICc, weights, and path coefficients validated
against phylopath::phylo_path() in R
(see tests/r_validation/validate_phylo_path.R).