Phylogenetic Ordination
Ordination incorporating phylogenetic structure (supports discordance-aware VCV with -g)
Command identity
- Canonical command:
phylogenetic_ordination- Handler:
phylogenetic_ordination- Aliases:
dimreduce, ord, ordination, pdr, phyl_pca, phylo_dimreduce, phylo_ordination, phylo_pca, ppca
- Standalone executables:
pk_phylogenetic_ordination, pk_dimreduce, pk_ord, pk_ordination, pk_pdr, pk_phyl_pca, pk_phylo_dimreduce, pk_phylo_ordination, pk_phylo_pca, pk_ppca
- Categories:
Phylogenetic comparative methods
Runtime interface
Synopsis
phykit phylogenetic_ordination --tree <tree> --trait_data <trait_data> [--method <method>] [--correction <correction>] [--mode <mode>] [--n-components <n_components>] [--perplexity <perplexity>] [--n-neighbors <n_neighbors>] [--min-dist <min_dist>] [--seed <seed>] [--plot] [--plot-tree] [--no-plot-tree] [--color-by <color_by>] [--tree-color-by <tree_color_by>] [--plot-output <plot_output>] [--gene-trees <gene_trees>] [--fig-width <fig_width>] [--fig-height <fig_height>] [--dpi <dpi>] [--no-title] [--title <title>] [--legend-position <legend_position>] [--ylabel-fontsize <ylabel_fontsize>] [--xlabel-fontsize <xlabel_fontsize>] [--title-fontsize <title_fontsize>] [--axis-fontsize <axis_fontsize>] [--colors <colors>] [--ladderize] [--cladogram] [--circular] [--color-file <color_file>] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
false |
str |
pca |
pca, tsne, umap |
|
false |
str |
BM |
BM, lambda |
|
false |
str |
cov |
cov, corr |
|
false |
int |
2 |
any |
|
false |
float |
none |
any |
|
false |
int |
none |
any |
|
false |
float |
0.1 |
any |
|
false |
int |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
str |
phylo_ordination_plot.png |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
int |
300 |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Output-file options: --plot-output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
Function names: phylogenetic_ordination; phylo_ordination; ordination; ord;
phylo_pca; phyl_pca; ppca; phylo_dimreduce; dimreduce; pdr
Command line interface: pk_phylogenetic_ordination; pk_phylo_ordination;
pk_ordination; pk_ord; pk_phylo_pca; pk_phyl_pca; pk_ppca;
pk_phylo_dimreduce; pk_dimreduce; pk_pdr
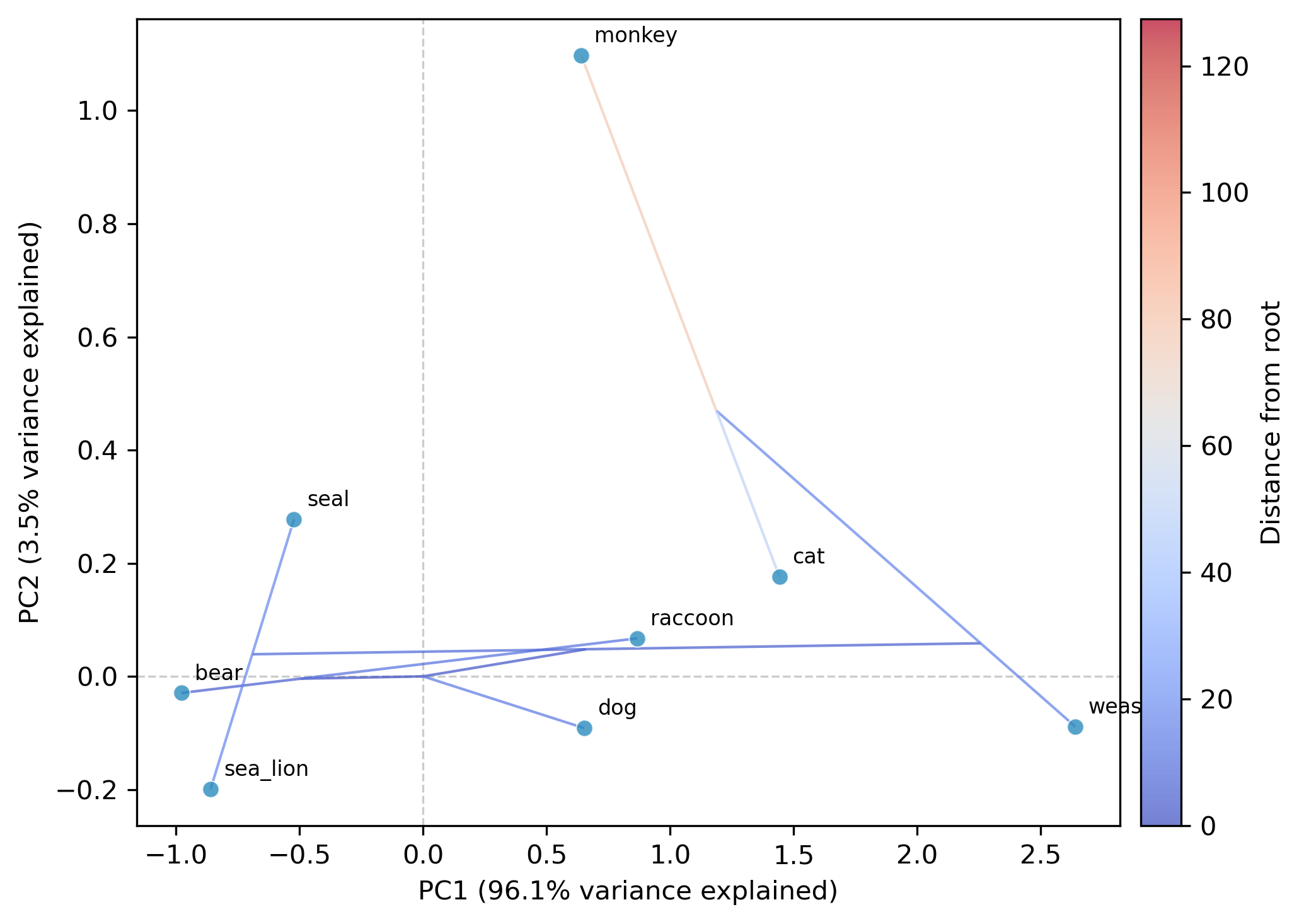
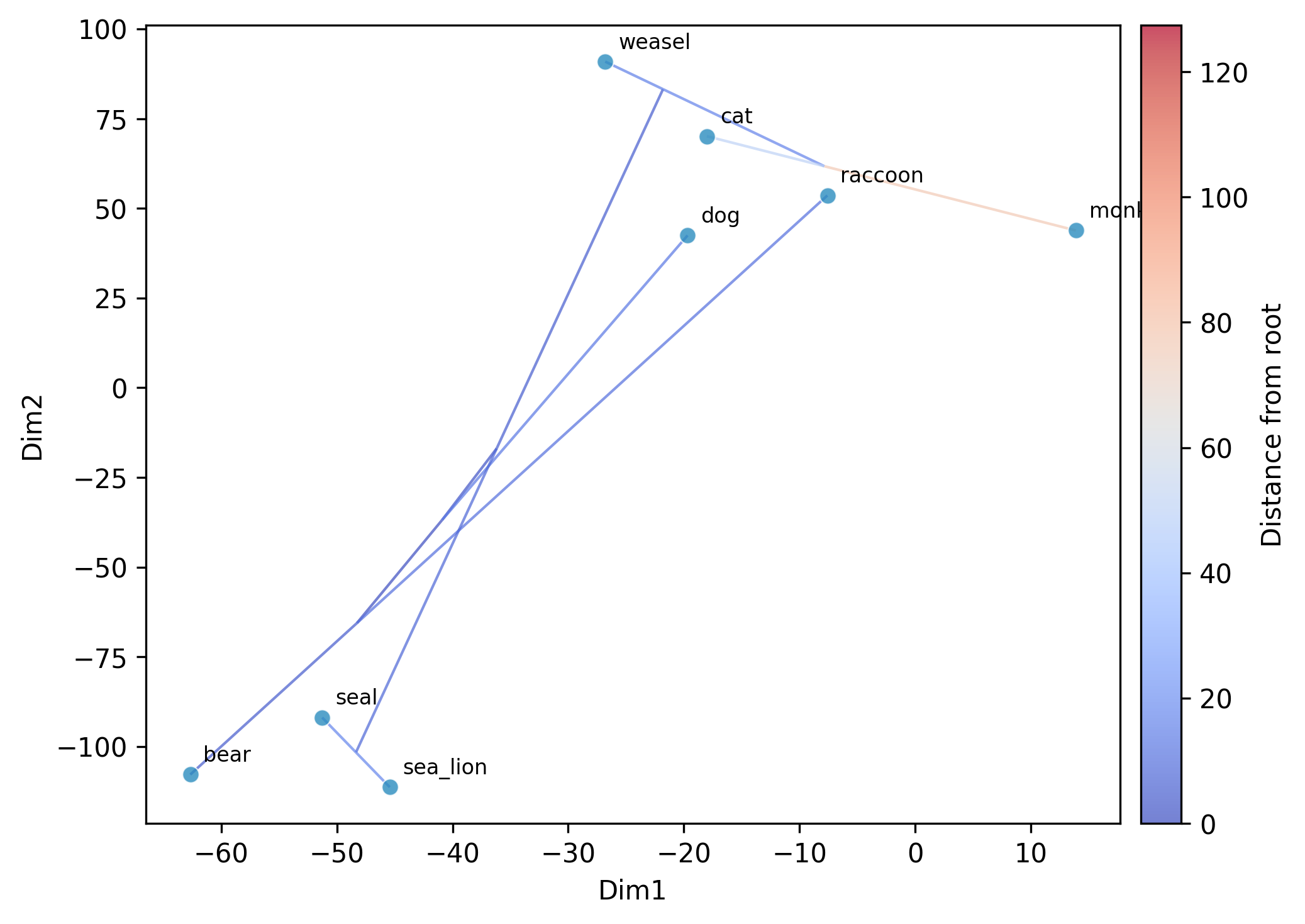
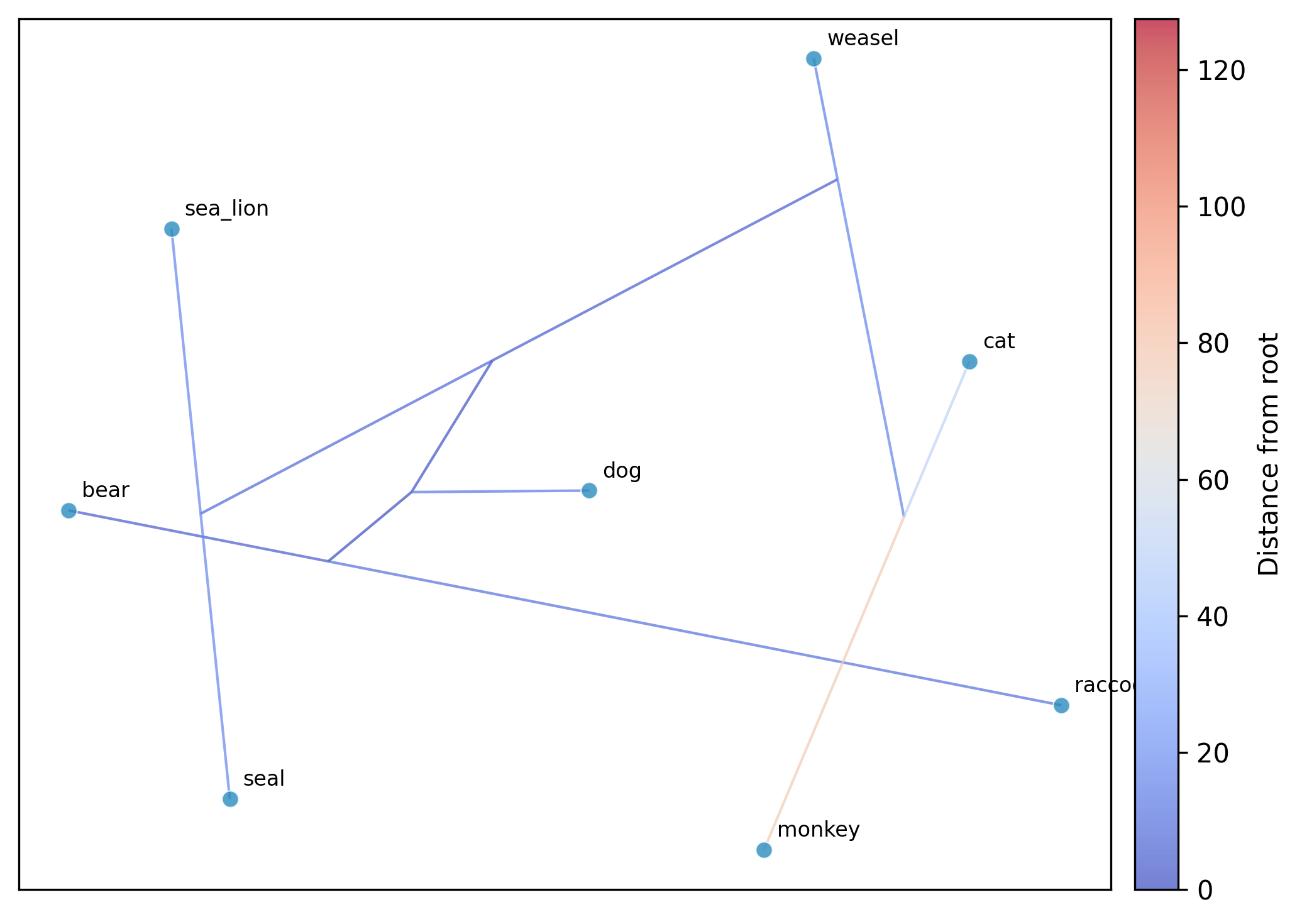
Perform phylogenetic ordination (PCA, t-SNE, or UMAP) on continuous multi-trait data while accounting for phylogenetic non-independence among species.
All methods use GLS-centering via the phylogenetic variance-covariance matrix. For PCA, eigendecomposition of the evolutionary rate matrix is performed to extract principal components. For t-SNE and UMAP, nonlinear embedding is applied to the GLS-centered data.
Three ordination methods are available via --method:
pca (default): phylogenetic PCA (Revell 2009). Performs eigendecomposition of the evolutionary rate matrix.
tsne: t-SNE embedding of the GLS-centered data. Perplexity is auto-set to
min(30, (n-1)/3); requires at least 4 taxa.umap: UMAP embedding of the GLS-centered data. n_neighbors is auto-set to
min(15, n-1); requires at least 3 taxa.
Two phylogenetic correction modes are available via --correction:
BM (default): assumes traits evolved under Brownian motion. The phylogenetic VCV matrix is used directly.
lambda: jointly estimates a single Pagel's lambda parameter across all traits by maximum likelihood, then rescales the off-diagonal elements of the VCV matrix. This accounts for deviations from Brownian motion.
For PCA, two modes are available via --mode:
cov (default): PCA on the evolutionary rate (covariance) matrix.
corr: PCA on the evolutionary rate correlation matrix, which standardizes traits to equal variance before extracting components.
The multi-trait input file should be tab-delimited with a header row:
taxon<tab>trait1<tab>trait2<tab>...
Lines starting with '#' are treated as comments. If the tree and trait file
have different taxa, the intersection is used and warnings are printed to
stderr.
PCA output includes eigenvalues with proportion of variance explained, trait
loadings, and taxon scores for each principal component. t-SNE/UMAP output
includes method parameters and embedding coordinates. When correction=lambda
is used, the estimated lambda and log-likelihood are also reported.
PCA results have been benchmarked against the R package
phytools
(phyl.pca function; Revell 2012). Eigenvalues, loadings, and scores match
phytools across all method/mode combinations (BM+cov, BM+corr, lambda+cov,
lambda+corr) within numerical tolerance (1e-4).
phykit phylogenetic_ordination -t <tree> -d <trait_data> [--method <pca|tsne|umap>] [--correction <BM|lambda>] [--mode <cov|corr>] [--n-components <int>] [--perplexity <float>] [--n-neighbors <int>] [--min-dist <float>] [--seed <int>] [--plot] [--plot-tree] [--no-plot-tree] [--color-by <col_or_file>] [--tree-color-by <col_or_file>] [--plot-output <path>] [-g <gene_trees>]
[--fig-width <float>] [--fig-height <float>] [--dpi <int>] [--no-title] [--title <str>]
[--legend-position <str>] [--ylabel-fontsize <float>] [--xlabel-fontsize <float>]
[--title-fontsize <float>] [--axis-fontsize <float>] [--colors <str>] [--ladderize] [--cladogram] [--circular] [--color-file <file>] [--json]
Options:
-t/--tree: a tree file in Newick format
-d/--trait_data: tab-delimited multi-trait file with header row
--method: ordination method: pca, tsne, or umap (default: pca)
--correction: phylogenetic correction: BM or lambda (default: BM)
--mode: PCA mode: cov or corr (default: cov; PCA only)
--n-components: number of embedding dimensions (default: 2; tsne/umap only)
--perplexity: t-SNE perplexity (default: auto)
--n-neighbors: UMAP n_neighbors (default: auto)
--min-dist: UMAP min_dist (default: 0.1)
--seed: random seed for reproducibility
--plot: optional argument to save a scatter plot
--plot-tree: overlay the phylogeny via ancestral reconstruction (default for tsne/umap; opt-in for pca)
--no-plot-tree: disable the phylogeny overlay for tsne/umap plots
--color-by: color tip points by a trait; specify a column name from the multi-trait file or a separate tab-delimited file (taxon<tab>value) for continuous or discrete coloring
--tree-color-by: color phylogeny edges by a trait; specify a column name or a tab-delimited file (default: distance from root)
--plot-output: output path for plot (default: phylo_ordination_plot.png)
-g/--gene-trees: optional multi-Newick file of gene trees; when provided, uses a discordance-aware VCV (genome-wide average) instead of the species-tree VCV
--fig-width: figure width in inches (auto-scaled if omitted)
--fig-height: figure height in inches (auto-scaled if omitted)
--dpi: resolution in DPI (default: 300)
--no-title: hide the plot title
--title: custom title text
--legend-position: legend location (e.g., "upper right", "none" to hide)
--ylabel-fontsize: font size for y-axis labels; 0 to hide
--xlabel-fontsize: font size for x-axis labels; 0 to hide
--title-fontsize: font size for the title
--axis-fontsize: font size for axis labels
--colors: comma-separated colors (hex or named)
--ladderize: ladderize (sort) the tree before plotting
--cladogram: draw cladogram (equal branch lengths, tips aligned) instead of phylogram
--circular: draw circular (radial/fan) phylogram instead of rectangular
--color-file: color annotation file for tip labels, clade ranges, and branch colors (iTOL-inspired TSV format)
--json: optional argument to print results as JSON