Inferring a phylogeny, from sequences to a tree
Introduction
Step 1: Data collection and software installation
1.1. Set up your working directory
1.2. Collect data
1.3. Install software
Step 2: Align and trim the sequences
2.1. Infer a multiple sequence alignment
2.2. Trim the multiple sequence alignment
Step 3: Reconstruct the evolutionary history of the sequences
Step 4: Automate steps 1-3 without writing any code!
Closing remarks
References
Introduction
Phylogenetics facilitates diverse downstream analyses .
Phylogenetics aims to infer the evolutionary relationships among biological features. Among other applications,
phylogenetics can facilitate organismal classification, orthology inference, and forensics.
Here, we will go over a ‘typical’ workflow from a collection of sequences to a phylogenetic tree using a maximum
likelihood framework. This tutorial does not aim to be exhaustive, but will go into greater detail for certain
steps in the workflow.
As noted in another tutorial I’ve written, titled Five-step phylogenomics, from proteomes
to species tree, this tutorial is grounded in statistics and will therefore make several assumptions.
Careful consideration of caveats and sources of error are worthwhile but beyond the scope of the tutorial.
Also, phylogenies represent hypotheses, not absolute ground truths. Differences in experimental design
(e.g., software choice) may impact inferences.
With that, let’s make a tree!
Step 1: Data collection and software installation
1.1. Set up your working directory
To keep everything organized, we will first make a directory for this tutorial called seqs_to_tree with the
mkdir command:
mkdir seqs_to_tree
.
Thereafter, move into the directory you made with the cd command:
cd seqs_to_tree
.
1.2. Collect data
Next, identify the sequenecs you are interested in constructing a phylogenetic tree for. These could be a
predicted orthologous group of genes, loci identified from sequence similarity searches, or products from
polymerase chain reactions. For ease of use, I have precompiled some sequneces of the gene
Prestin,
a cochlear motor protein, from Humans and close relatives.
Move the downloaded sequences to the current working directory with the mv command:
mv path_to_file/prestin.fa .
. Be sure to change path_to_file to the appropriate relative or
absolute file path of prestin.fa.
1.3. Install software
Downloading and installing software is left out of most tutorials, but it is an essential step. Accordingly, we will go over it. Some software we will use can be downloaded from PyPi. To do so, we will continue to implement best practices and first create a virtual environment to store all installed software. A virtual environment is a "self-contained directory tree that contains a Python installation for a particular version of Python, plus a number of additional packages" (Python docs). Create a virtual environment with the following command:
python -m venv venv
and activate it
source venv/bin/activate
.
Next, install
ClipKIT,
PhyKIT, and
BioKIT using the pip install command:
pip install clipkit jlsteenwyk-biokit
.
Disclaimer: these are all software I have developed. I developed these software
and distribute them freely to empower and enable bioinformaticians of varying experience.
I hope you find them helpful :D
The last pieces of software that need to be installed are MAFFT and IQ-TREE. Each of these software have their own installation instructions, which are better than what I could provide. Please follow the link for each software and install them according to the developer's recommendations. Add the path of each executable to your $PATH variable. Instructions to do so can be found on numerous online discussion forums such as askubuntu and unix stackexchange.
Step 2: Align and trim the sequences
2.1. Infer a multiple sequence alignment
Algorithms that infer multiple sequence alignments attempt to identify site-wise homology among loci.
Sites that are homologous are represented as columns in the multiple sequence alignment.
First, let's inspect the contents of prestin.fa. For example, we could examine the FASTA headers
of each sequence represented in the FASTA file. To do so, use the grep command:
grep ">" prestin.fa
>Bat [Pteropus vampyrus]
>Bat [Rousettus aegyptiacus]
>Bat [Pteropus alecto]
>Bat [Desmodus rotundus]
>Bat [Molossus molossus]
>Whale [Balaenoptera acutorostrata scammoni]
>Dolphin [Tursiops truncatus]
>Dolphin [Lagenorhynchus obliquidens]
>Human [Homo sapiens]
>Orangutan [Pongo abelii]
>Gorilla [Gorilla gorilla gorilla]
>Squirrel [Neosciurus carolinensis]
>Dog [Canis lupus familiaris]
>Dingo [Canis lupus dingo]
grep ">" prestin.fa -c
. There are 14 sequences.
To align the sequences, use MAFFT with the "auto" parameter (Katoh,K. and Standley, 2013) by executing the following command:
mafft --auto prestin.fa > prestin.fa.mafft. Take a look at the contents of the
file using the head command. Gap characters (represented as dashes "-") have been added to the file. Gaps may reflect sites that have
undergone histories of insertions and deletions.
2.2. Trim the multiple sequence alignment
To trim sequences, we will use ClipKIT with the default "smart-gap" mode, which dynamically infers a gap threshold for trimming; in other words, the gap-rich sites, which are poor in phylogenetic information, will be removed from the alignment (Steenwyk et al., 2020). Trim the alignment using the following command:
clipkit prestin.fa.mafft. Lots of information will be printed to the screen and an output file
with the suffix ".clipkit" will have been generated.
Examine the features of the multiple sequence alignment using BioKIT; specifically, the aln_summary function.
biokit aln_summary prestin.fa.mafft.clipkit
General Characteristics
=======================
14 Number of taxa
712 Alignment length
64 Parsimony informative sites
64 Variable sites
648 Constant sites
Character Frequencies
=====================
Y 391
W 32
V 944
T 591
S 603
R 289
Q 321
P 530
N 370
M 226
L 1072
K 501
I 724
H 149
G 761
F 544
E 518
D 409
C 126
A 840
- 27Step 3: Reconstruct the evolutionary history of the sequences
3.1. Infer the phylogenetic tree using IQ-TREE
To infer the evolutionary history of the sequences, we will use the maximum likelihood approach implemented
in IQ-TREE
(Minh et al., 2020).
To do so, execute the following command:
iqtree2 -s prestin.fa.mafft.clipkit -m TEST -bb 5000
.
Under the hood, the best fitting substitution model is automatically selected using
ModelFinder
(Kalyaanamoorthy et al., 2017).
Using the "-bb" argument, 5,000 ultrafast bootstrap approximations will be used to evaluate bipartition support
(Hoang et al., 2017).
IQ-TREE generates many useful files, however, we are most
interested in the best tree identified during tree search, which is prestin.fa.mafft.clipkit.treefile.
To obtain a quick view of the inferred phylogeny, we will use the "print_tree" function in
PhyKIT by executing the following command:
phykit print_tree prestin.fa.mafft.clipkit.treefile
, Bat__Pteropus_vampyrus_
|
|____ Bat__Rousettus_aegyptiacus_
|
, Bat__Pteropus_alecto_
_|
| __________ Bat__Desmodus_rotundus_
| ____|
| | |_________ Bat__Molossus_molossus_
| |
| | , Human__Homo_sapiens_
| _| |
| | | ________| Gorilla__Gorilla_gorilla
| | | | |
| | | ____| | Orangutan__Pongo_abelii_
| | | | |
| | |________| |_______ Squirrel__Neosciurus_carolinensis_
|_______| |
| | , Dog__Canis_lupus
| |_______|
| | Dingo__Canis_lupus
|
| _________ Whale__Balaenoptera_acutorostrata
|___________|
| , Dolphin__Tursiops_truncatus_
|_________________|
| Dolphin__Lagenorhynchus_obliquidens_
Step 4: Automate steps 1-3 without writing any code!
I recognize that the command line is not for everyone. To this end, I developed a workflow
on the Latch console and published it to the following dedicated link:
https://console.latch.bio/explore/62302/info.
(As of August 2022, I do a few hours of consulting work with Latch a month. I note this to be transparent about any potential
conflict of interest.) Users can make accounts for free on the Latch platform Latch and the
workflow can also be run for free. When you open the web link, you
will see something like the following:
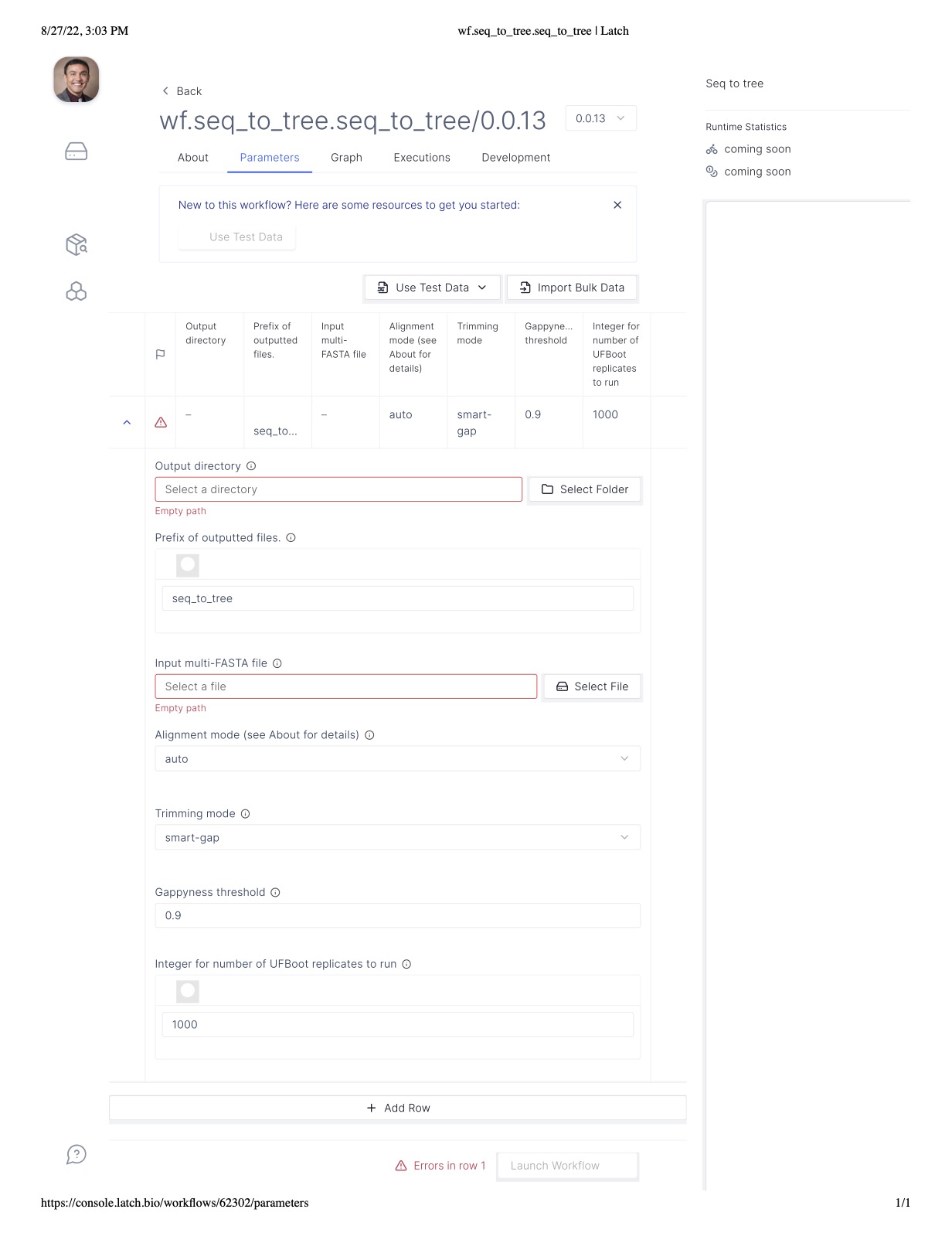 Simply fill in the required boxes (shown in red), modify optional arguments such as the alignment
and trimming strategies as you see fit, and launch the workflow! The workflow will automatically conduct
steps 1 through 3 of this tutorial and save all intermediate and output files.
Simply fill in the required boxes (shown in red), modify optional arguments such as the alignment
and trimming strategies as you see fit, and launch the workflow! The workflow will automatically conduct
steps 1 through 3 of this tutorial and save all intermediate and output files.
Closing remarks
This tutorial covered how to infer a phylogenetic tree (and a little more!) in three steps.
The resulting phylogenetic tree can be used for diverse downstream analyses, facilitate hypothesis
generation, or be used as an endpoint in an analytical workflow.
Congratulations on inferring a phylogenetic tree! Please do not hesitate to
contact me if you have any questions or constructive comments.
References
1. Hoang,D.T. et al. (2018)
UFBoot2: Improving the
Ultrafast Bootstrap Approximation. Mol. Biol. Evol., 35, 518–522.
2. Kalyaanamoorthy,S. et al. (2017)
ModelFinder: fast model selection
for accurate phylogenetic estimates. Nat. Methods, 14, 587–589.
3. Minh,B.Q. et al. (2020)
IQ-TREE 2: New Models
and Efficient Methods for Phylogenetic Inference in the Genomic Era.
Mol. Biol. Evol., 37, 1530–1534.
4. Steenwyk,J.L., et al. (2021)
BioKIT: a versatile
toolkit for processing and analyzing diverse types of sequence data. Genetics,
10.1093/genetics/iyac079.
5. Steenwyk,J.L. et al. (2020)
ClipKIT:
A multiple sequence alignment trimming software for accurate phylogenomic inference.
PLOS Biol., 18, e3001007.
6. Steenwyk,J.L., et al. (2021)
PhyKIT:
a broadly applicable UNIX shell toolkit for processing and analyzing phylogenomic data.
Bioinformatics.