Character map (synapomorphy/homoplasy mapping)
Map synapomorphies and homoplasies onto a phylogeny
Command identity
- Canonical command:
character_map- Handler:
character_map- Aliases:
charmap, synapomorphy_map
- Standalone executables:
pk_character_map, pk_charmap, pk_synapomorphy_map
- Categories:
Trait evolution
Runtime interface
Synopsis
phykit character_map --tree <tree> --data <data> --output <output> [--optimization <optimization>] [--phylogram] [--characters <characters>] [--allow-taxon-mismatch] [--change-marker-size <change_marker_size>] [--change-fontsize <change_fontsize>] [--verbose] [--fig-width <fig_width>] [--fig-height <fig_height>] [--dpi <dpi>] [--no-title] [--title <title>] [--legend-position <legend_position>] [--ylabel-fontsize <ylabel_fontsize>] [--xlabel-fontsize <xlabel_fontsize>] [--title-fontsize <title_fontsize>] [--axis-fontsize <axis_fontsize>] [--colors <colors>] [--ladderize] [--cladogram] [--circular] [--color-file <color_file>] [--json]
Arguments
This table is generated from the live command parser. It is the authoritative source for accepted spellings, required arguments, types, defaults, and choices.
Argument |
Required |
Type |
Default |
Choices |
|---|---|---|---|---|
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
true |
str |
required |
any |
|
false |
str |
acctran |
acctran, deltran |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
boolean |
false |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
int |
300 |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
str |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
float |
none |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
boolean |
false |
any |
|
false |
str |
none |
any |
|
false |
boolean |
false |
any |
Output and errors
--json provides the command's structured JSON representation. Output-file options: --output. Invalid command syntax exits with status 2. Input
validation and scientific limitations are described in the guidance below.
Guidance, interpretation, and examples
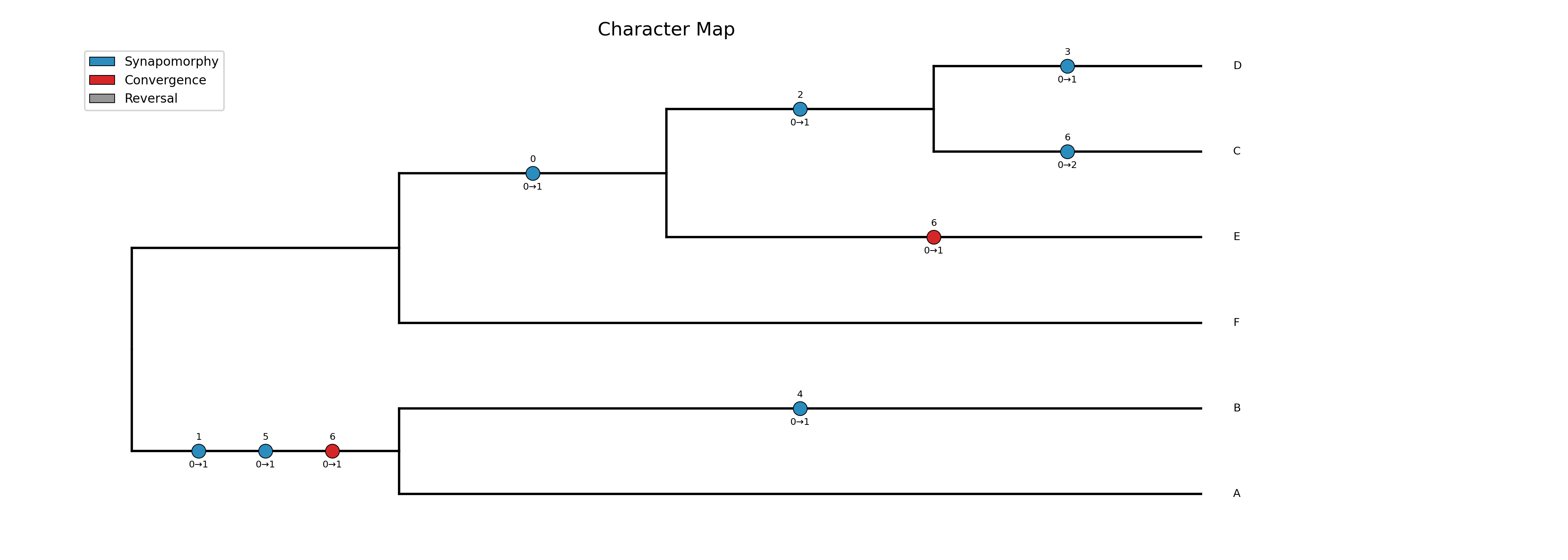
Map character state changes onto a phylogeny using unordered parsimony with ACCTRAN (default) or DELTRAN optimization. Produces a cladogram (default) or phylogram with color-coded circles on each branch showing where character state changes occur. This is useful for visualizing synapomorphies and homoplasies in morphological datasets, similar to the classic Winclada software.
Each circle on a branch represents a character state change. The
number above the circle is the character index (matching the column
in the input matrix). The transition below shows the old and new
state (e.g., 0→1). Circle color indicates the type of change:
Blue circles: synapomorphies — this character changed to this state only once on the entire tree, uniquely supporting the clade
Red circles: convergences — this character independently gained the same state on two or more branches
Gray circles: reversals — the character returned to a state previously seen at an ancestor
Note that the same state transition (e.g., 0→1) may appear in
different colors because the color reflects how many times that
particular character underwent that transition across the tree, not the
state values themselves.
Input: a Newick tree file and a TSV character matrix (header row with
character names, one row per taxon with discrete states). Missing data
(? or -) is treated as a wildcard.
Reports the consistency index (CI), retention index (RI), and total tree length (parsimony steps). CI and RI cross-validated against R's phangorn.
Polytomies are retained in the analysis and plot. Character states and parsimony scores on multifurcating trees are calculated directly on the submitted unresolved topology rather than on an arbitrary binary resolution.
Taxon labels in the tree and character matrix must match exactly and are
case-sensitive. By default, any label found in only one input is reported and
the command exits without producing a character map. Use
--allow-taxon-mismatch to warn and analyze only the shared taxa instead.
Every tree tip and matrix row must have a unique, non-empty taxon label.
Example usage:
# Basic usage with default ACCTRAN optimization and cladogram layout
phykit character_map -t species.tre -d morphology.tsv -o charmap.png
# Use DELTRAN optimization and ladderize the tree
phykit character_map -t species.tre -d morphology.tsv -o charmap.png \
--optimization deltran --ladderize
# Show only specific characters of interest
phykit character_map -t species.tre -d morphology.tsv -o charmap.png \
--characters 0,3,7,12
# Phylogram layout with JSON output
phykit character_map -t species.tre -d morphology.tsv -o charmap.png \
--phylogram --json
Input format — the character matrix is a tab-separated file where the first row is a header with character names, and each subsequent row has a taxon name followed by the character states:
taxon char0 char1 char2 char3
Taxon_A 0 1 0 2
Taxon_B 0 1 1 0
Taxon_C 1 0 0 2
Taxon_D 1 0 1 1
Taxon labels are not normalized: leading or trailing whitespace in the TSV is
retained and therefore affects matching. In Newick files, labels containing
spaces or reserved punctuation should be enclosed in single quotes, for
example 'Taxon one'. The corresponding TSV label is Taxon one without
the quote characters.
Full usage:
phykit character_map -t <tree> -d <data> -o <output>
[--optimization acctran|deltran] [--phylogram]
[--characters 0,1,3] [--allow-taxon-mismatch]
[--change-marker-size <float>] [--change-fontsize <float>]
[--verbose] [--json]
[--fig-width <float>] [--fig-height <float>] [--dpi <int>] [--no-title] [--title <str>]
[--legend-position <str>] [--ylabel-fontsize <float>] [--xlabel-fontsize <float>]
[--title-fontsize <float>] [--axis-fontsize <float>] [--colors <str>] [--ladderize]
Options:
-t/--tree: tree file in Newick format (required)
-d/--data: TSV character matrix with header row (required)
-o/--output: output figure path (.png, .pdf, .svg) (required)
--optimization: ancestral state optimization: acctran (default) or deltran
--phylogram: draw phylogram instead of cladogram
--characters: comma-separated character indices to display (0-based; all characters are still used for CI/RI)
--allow-taxon-mismatch: warn and analyze only taxa shared by the tree and matrix; mismatches are errors by default
--change-marker-size: positive character-change circle size in points squared; automatically scaled when omitted
--change-fontsize: positive font size for character indices and state transitions; automatically scaled when omitted
--verbose: print per-character detail
--colors: comma-separated colors for synapomorphy, convergence, reversal (default: blue, red, gray)
--ladderize: ladderize (sort) the tree before plotting
--cladogram: draw cladogram (equal branch lengths, tips aligned) instead of phylogram
--circular: draw circular (radial/fan) phylogram instead of rectangular
--color-file: color annotation file for tip labels, clade ranges, and branch colors (iTOL-inspired TSV format)
--json: optional argument to print results as JSON
R validation: Validated against phytools in R
(see tests/r_validation/validate_character_map.R).