Sensitivity analysis: detecting incongruence at different scales
Introduction
Objective, relevant literature and tutorial technicalities
Section 1: Data collection and preparation
1.1. Set up your working directory
1.2. Collect data
1.3. Examine dataset
Section 2: Phylogenomic subsampling across genes
2.1. Summarize information content across genes
2.2. Visualize information content from genes
2.3. Conduct sensitivity analysis
Section 3: Phylogenomic subsampling across taxa
3.1. Summarize information content across taxa
3.2. Visualize information content from taxa
3.3. Create subsampled data matrices and infer phylogenies
3.4. Determine the impact of subsampling taxa on species tree inference
Section 4: Phylogenomic subsampling across sites
4.1. Summarize information content across sites
4.2. Infer phylogenies from subsampled data
4.3. Compare subsampled inferred trees to the species tree
Closing remarks
Summary
Additional reading
Acknowledgements
Thank you!
References
Introduction
The objective of sensitivity analysis
Phylogenomics provides the necessary framework for conducting comparative studies, tracing the origin of phenotypes,
and pinpointing genomic innovation and loss. However, accurately inferring organismal histories is not trivial.
Sensitivity analysis plays a crucial role in phylogenomics, serving as a valuable method for assessing the robustness of
evolutionary relationships.
This approach is particularly important for:
By systematically varying input parameters and data subsets, sensitivity analysis can reveal branches or clades that are susceptible to changes in the underlying data or analytical methods.
It helps researchers pinpoint areas of the phylogeny where different genes, taxa or sites that have conflicting evolutionary signals.
Unstable branches highlighted by sensitivity analysis often warrant deeper examination, as they may indicate interesting biological phenomena (e.g., incomplete lineage sorting and introgression/hybridization).
In summary, sensitivity analysis enables researchers to test the reliability of phylogenomic inference and potentially uncover biological processes influencing organismal history.
Additional reading
In this tutorial, we will go over methods to conduct sensitivity analysis at the scale of genes, sites, and taxa. This tutorial covers many facets of phylogenomic sensitivity analysis, but, like any tutorial, it is not exhaustive. For additional reading on phylogenomic incongruence and various sources of error, see (Steenwyk et al., 2023, Nature Reviews Genetics).
As is true of any analysis grounded in statistics, assumptions will be made. An in-depth understanding of the assumptions being made during phylogenomic analysis is important, but that is beyond the scope of this tutorial. Readers may refer to an non-exhaustive list of additional resources for further reading to enhance their understanding of potential caveats and sources of error — e.g., Kapli et al. (2020), Nature Reviews Genetics; Yang & Rannala (2012), Nature Reviews Genetics; and Philippe et al. (2005), Annual Review of Ecology, Evolution, and Systematics.
I'd also like to acknowledge that species trees are hypotheses, not ground truths.
Technicalities of this tutorial
This tutorial assumes some familiarity with basic UNIX commands such as making directories, changing into them, etc. This tutorial will also assume familiarity with the R programming language. Commands in the UNIX or R prgrogramming environment will be displayed as code blocks that have a light grey background. For example,
this is a command you would execute!
This tutorial also extensively uses PhyKIT, a multi-tool for phylogenomics that I engineered (Steenwyk et al. 2021, Bioinformatics). PhyKIT is a tool for phylogenomics, not the tool for phylogenomics. So, while this tutorial frequently will use PhyKIT, many other great tools exist and I encourage you to check them out and use what fits your needs and style.
Throughout the tutorial, hints and solutions will be provided. Hints and solutions will look as follows and to access them, simply click on the word "Hint" or the word "Solution":
Hint
This is a hint!
Solution
This is a solution!
Lastly, I also want to acknowledge a source of phylogenomic error that we may encounter during this tutorial. Specifically, reproducibility in phylogenomics. It seems hard to believe but even using the same seed during tree search does not guarantee inferring the same phylogeny. In fact, this is observed among around 9-18% of single-locus phylogenetic trees (Shen et al. 2020, Nature Communications). This can be due to several reasons, some of which involve how hardware interacts with software and are beyond the scope of the present tutorial. However, if you notice slight differences between your results and the results presented here, there is a non-zero possibility that it is driven by irreproducibility.
With that, let's do some phylogenomic sensitivity analysis!
Section 1: Data collection and preparation
Phylogenomic subsampling first requires that researchers have assembled a dataset.
The objective of this step is to assemble and prepare the dataset for downstream
alignment and single-gene tree inference. The resulting alignments and single-gene
trees will be further processed in the sensitivity/subsampling analysis.
1.1. Set up your working directory
To keep everything organized, we will first make a directory for this tutorial called sensitivity_analysis with the mkdir command.
Solution
mkdir sensitivity_analysis
Thereafter, move into the directory you made with the cd command.
Solution
cd sensitivity_analysis
1.2. Collect data
1) I have precompiled a set of trimmed alignments and single-gene phylogenies for the purposes of this tutorial.
While phylogenomics typically involves several hundreds of genes,
we will focus on two sets of 50 orthologs each — one set from mammals and another from yeast. Mammalian sequences
were obtained from Ensembl BioMart; yeast sequences were obtained from the
Yeast Gene Order Browser.
Click these links to download the data or use wget or curl.
Solution: wget or curl
# curl solution
curl -O https://jlsteenwyk.com/tutorials/ub_sensitivity_files/data_mammals.tar.gz
curl -O https://jlsteenwyk.com/tutorials/ub_sensitivity_files/data_yeast.tar.gz
# wget solution
wget https://jlsteenwyk.com/tutorials/ub_sensitivity_files/data_mammals.tar.gz
wget https://jlsteenwyk.com/tutorials/ub_sensitivity_files/data_yeast.tar.gz
2) Unzip the directories with the tar command.
Note, before doing so, double check that the downloaded files are in the correct directory. They should be in your current working directory sensitivity_analysis. If they are not, use the mv command to move them into your current directory.
Solution
tar -zxvf data_mammals.tar.gz ;
tar -zxvf data_yeast.tar.gz
1.3. Examine dataset
1) Look at the contents of each directory and determine what information is in each file/subdirectory.
Solution
ls data_mammals
FILES_trees FILES_trimmed Mammals_2002_eMRC.tree
# EXPLANATION:
# FILES_trees: single-locus phylogenies that have been already been inferred to save time
# FILES_trimmed: trimmed multiple sequence alignments. Sequences were aligned using MAFFT and trimmed using ClipKIT with default parameters.
# Mammals_2002_eMRC.tree: Species tree inferred using several thousand loci. Only a subset of loci are used in this tutorial for the sake of time.
The same set of files and naming convention are observed in the data_yeast directory.
2) Examine the names of the tree tips and FASTA entries. Do they match?
Hint
All alignments have 100% taxon occupancy, meaning all species have an ortholog for each gene.
As a result, we can take a look at the names in any alignment file (e.g., data_mammals/FILES_unaligned/24_ENSG00000000005.fasta)
and compare them to the tree file and see if they match.
For getting the names of the tips in the phylogeny, use the pk_labels command in PhyKIT.
Solution
paste <(pk_labels data_mammals/Mammals_2002_eMRC.tree | sort) <(grep ">" data_mammals/FILES_trimmed/24_ENSG00000000005.fasta.mafft.clipkit | sed 's/>//g' | sort)
# EXPLANATION:
# Yes, the FASTA entry names match the tree name.
# It is helpful to set up files this way because everything downstream is easier.
# The same set of files and naming convention are observed in the data_yeast directory.
Section 2: Phylogenomic subsampling across genes
The objective of this step is to calculate diverse measures of information content of the phylogenomic data matrices.
2.1. Summarize information content across genes
1) There are numerous metrics that capture the information content of multiple sequence alignments and phylogenetic trees. Here, we will calculate
seven of the many metrics. Specifically, we will calculate:
2) For each ortholog, calculate all seven metrics that capture many features of information content using PhyKIT. Save the output to a file titled info_content_genes.txt. The output should have four columns.
Note, this step will take some time. Feel free to stretch your legs and check on how your neighbors are progressing. If you are not familiar with nested for loops, we will be using them throughout the tutorial. As a result, it would be good to make sure you understand the nested for loop solution.
Hint
Links to PhyKIT documentation for each metric are provided in the text above.
There are two solutions. One uses a nested for loop and the other does not.
If you are familiar with loops, but not nested loops, compare the two solutions
to better understand how a nested for loop works. They will be used extensively
throughout the remainder of the tutorial.
Solution: nested for loop
for dataset in mammals yeast;
do
for i in $(ls data_${dataset}/FILES_trimmed/);
do
aln=$(echo "$i")
tre=$(echo "${i}.treefile")
id=$(echo "$i" | sed 's/.fa.*$//g')
aln_len=$(pk_aln_len data_${dataset}/FILES_trimmed/${aln})
abs=$(pk_bss data_${dataset}/FILES_trees/${tre} | grep "mean" | awk '{print $NF}')
rcv=$(pk_rcv data_${dataset}/FILES_trimmed/${aln})
lbs=$(pk_lbs data_${dataset}/FILES_trees/${tre} | grep "median" | awk '{print $NF}')
tness=$(pk_tness data_${dataset}/FILES_trees/${tre})
sat=$(pk_sat -a data_${dataset}/FILES_trimmed/${aln} -t data_${dataset}/FILES_trees/${tre} | awk '{print $2}')
tor=$(pk_tor -a data_${dataset}/FILES_trimmed/${aln} -t data_${dataset}/FILES_trees/${tre} | awk '{print $1}')
echo -e "${dataset}\t${id}\taln_len\t$aln_len"
echo -e "${dataset}\t${id}\trcv\t$rcv"
echo -e "${dataset}\t${id}\tabs\t$abs"
echo -e "${dataset}\t${id}\tlbs\t$lbs"
echo -e "${dataset}\t${id}\ttreeness\t$tness"
echo -e "${dataset}\t${id}\tsaturation\t$sat"
echo -e "${dataset}\t${id}\ttreeness_over_rcv\t$tor"
done
done |tee info_content_genes.txt
Solution: no nested for loop
for i in $(ls data_mammals/FILES_trimmed/);
do
aln=$(echo "$i")
tre=$(echo "${i}.treefile")
id=$(echo "$i" | sed 's/.fa.*$//g')
aln_len=$(pk_aln_len data_mammals/FILES_trimmed/${aln})
abs=$(pk_bss data_mammals/FILES_trees/${tre} | grep "mean" | awk '{print $NF}')
rcv=$(pk_rcv data_mammals/FILES_trimmed/${aln})
lbs=$(pk_lbs data_mammals/FILES_trees/${tre} | grep "median" | awk '{print $NF}')
tness=$(pk_tness data_mammals/FILES_trees/${tre})
sat=$(pk_sat -a data_mammals/FILES_trimmed/${aln} -t data_mammals/FILES_trees/${tre} | awk '{print $2}')
tor=$(pk_tor -a data_mammals/FILES_trimmed/${aln} -t data_mammals/FILES_trees/${tre} | awk '{print $1}')
echo -e "mammals\t${id}\taln_len\t$aln_len"
echo -e "mammals\t${id}\trcv\t$rcv"
echo -e "mammals\t${id}\tabs\t$abs"
echo -e "mammals\t${id}\tlbs\t$lbs"
echo -e "mammals\t${id}\ttreeness\t$tness"
echo -e "mammals\t${id}\tsaturation\t$sat"
echo -e "mammals\t${id}\ttreeness_over_rcv\t$tor"
done |tee info_content_genes.txt
for i in $(ls data_yeast/FILES_trimmed/);
do
aln=$(echo "$i")
tre=$(echo "${i}.treefile")
id=$(echo "$i" | sed 's/.fa.*$//g')
aln_len=$(pk_aln_len data_yeast/FILES_trimmed/${aln})
abs=$(pk_bss data_yeast/FILES_trees/${tre} | grep "mean" | awk '{print $NF}')
rcv=$(pk_rcv data_yeast/FILES_trimmed/${aln})
lbs=$(pk_lbs data_yeast/FILES_trees/${tre} | grep "median" | awk '{print $NF}')
tness=$(pk_tness data_yeast/FILES_trees/${tre})
sat=$(pk_sat -a data_yeast/FILES_trimmed/${aln} -t data_yeast/FILES_trees/${tre} | awk '{print $2}')
tor=$(pk_tor -a data_yeast/FILES_trimmed/${aln} -t data_yeast/FILES_trees/${tre} | awk '{print $1}')
echo -e "yeast\t${id}\taln_len\t$aln_len"
echo -e "yeast\t${id}\trcv\t$rcv"
echo -e "yeast\t${id}\tabs\t$abs"
echo -e "yeast\t${id}\tlbs\t$lbs"
echo -e "yeast\t${id}\ttreeness\t$tness"
echo -e "yeast\t${id}\tsaturation\t$sat"
echo -e "yeast\t${id}\ttreeness_over_rcv\t$tor"
done |tee -a info_content_genes.txt
3) Determine if the mammal or yeast dataset has a higher or lower average RCV score. What is the interpretation for the dataset with a lower RCV score?
Hint
One solution is to write a loop that will loop through "mammals" and "yeast", obtain rows that contain those strings and "rcv"
and then calculate the average of the fourth column using awk.
Solution
for i in $(echo "mammals yeast"); do
avg_rcv=$(cat info_content_genes.txt | grep $i | grep rcv | awk '{s+=$4} END {print s/NR}') ;
echo -e "${i}\t${avg_rcv}" ;
done
mammals 0.659387
yeast 0.918839
# EXPLANATION:
Because the average RCV value is lower in the mammal dataset, the mammalian dataset is less susceptible to compositional biases.
4) Determine which dataset — the yeast or mammal dataset — is subject to higher amounts of saturation. What is the interpretation for the dataset with a lower saturation score?
Hint
The solution from the previous question can be tweaked to address this question.
Solution
for i in $(echo "mammals yeast"); do
res=$(cat info_content_genes.txt | grep $i | grep saturation | awk '{s+=$4} END {print s/NR}') ;
echo -e "$i\t$res" ;
done
mammals 0.227978
yeast 0.688004
# EXPLANATION:
Because the average value is lower in the mammal dataset, the mammalian dataset has fewer signatures of saturation.
5) For each dataset, what three genes have the best score for treeness / RCV?
Hint
Modify the previous solution and use the sort function in UNIX.
Also, higher values are better.
Solution
for i in $(echo "mammals yeast"); do
grep "$i" info_content_genes.txt | grep treeness_over_rcv | sort -k 4,4 -nr | head -n 3 ;
done
mammals 24_ENSG00000000005 treeness_over_rcv 2.0594
mammals 24_ENSG00000004534 treeness_over_rcv 2.0561
mammals 24_ENSG00000004799 treeness_over_rcv 1.8995
yeast 12_YAL012W_Anc_7.97 treeness_over_rcv 4.8591
yeast 12_YBL015W_Anc_8.161 treeness_over_rcv 3.9672
yeast 12_YAL026C_Anc_7.72 treeness_over_rcv 3.6659
# EXPLANATION:
For the mammals dataset,
24_ENSG00000000005, 24_ENSG00000004534, and 24_ENSG00000004799 have the highest signal to noise ratios.
For the yeast dataset,
12_YAL012W_Anc_7.97, 12_YBL015W_Anc_8.161, and 12_YAL026C_Anc_7.72 have the highest signal to noise ratios.
Moreover, these values indicate that the highest relative signal to noise ratios are observed in the yeast dataset.
6) For each dataset, what three genes have the best long branch score values?
Hint
Modify the previous solution and use the sort function in UNIX.
Also, lower values are better.
Solution
for i in $(echo "mammals yeast"); do
grep "$i" info_content_genes.txt | grep lbs | sort -k 4,4 -n | head -n 3 ;
done
mammals 24_ENSG00000000005 lbs -25.8322
mammals 24_ENSG00000006125 lbs -25.3831
mammals 24_ENSG00000008869 lbs -24.9082
yeast 12_YAL010C_Anc_7.103 lbs -21.191
yeast 12_YAR008W_Anc_4.116 lbs -20.786
yeast 12_YAL046C_Anc_7.23 lbs -20.7543
# EXPLANATION:
For the mammals dataset,
24_ENSG00000000005, 24_ENSG00000006125, and 24_ENSG00000008869 have the lowest long branch score values.
For the yeast dataset,
12_YAL010C_Anc_7.103, 12_YAR008W_Anc_4.116, and 12_YAL046C_Anc_7.23 have the lowest long branch score values.
2.2. Visualize information content from genes
The objective here is to learn how to make (near) publication-ready figures that summarize the distributions of the information content in both data matrices. Moreover,
Using R, Python, or any other method for plotting, plot the distribution of each metric. Note, the solution will be in the R programming language since many scientists are familiar with it and there is extensive, easily accessible documentation for plotting.
Hint
The R package ggplot2 can be very helpful for plotting. The facet_wrap() function will be very helpful for this data structure (i.e., long format).
Solution
# load R packages
library(ggplot2)
library(ggpubfigs)
# read in the data
dat<-read.table("info_content_genes.txt")
# rename the columns
colnames(dat) <- c("dataset", "gene", "metric", "value")
# use ggplot2 and ggpubfigs to make the figure
ggplot(dat, aes(dataset, value, fill=dataset)) +
geom_violin() + geom_boxplot(width=0.2, fill="white", alpha=0.7) +
facet_wrap(~metric, scales="free_y") + theme_simple() +
scale_fill_manual(values = friendly_pal("ito_seven"))
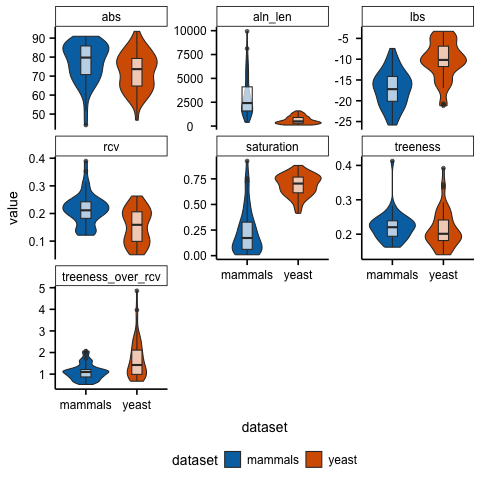
2.3. Conduct sensitivity analysis
1) Next, we will conduct sensitivity analysis for each dataset separately. The objective is to determine if any branches in the organismal history is unstable. Each metric accounts for different potential sources of error. For example, incongruence detected among genes with the best long branch scores suggests the organismal history may be subject to long branch attraction artifacts.
Create concatenation matrices for the top 50% best scoring genes for each metric using the create_concat function in PhyKIT.
Hint
There are 50 genes in each dataset. So, create concatenation matrices of the best scoring 25 genes.
Take a look at the help message for the create_concat function in PhyKIT:
pk_create_concat -h
Here is a summary of each of the metrics as well as if higher or lower values are better.
abs: higher is better
aln_len: higher is better
lbs: lower is better
rcv: lower is better
saturation: lower is better
treeness: higher is better
treeness_over_rcv: higher is better
Solution
for dataset in mammals yeast ; do
for metric in aln_len rcv abs lbs treeness saturation treeness_over_rcv ; do
metric_dat=$(grep "$dataset" info_content_genes.txt | grep "$metric") ;
echo "$dataset $metric"
if [[ "$metric" == "lbs" || "$metric" == "rcv" || "$metric" == "saturation" ]]; then
echo "$metric_dat" | sort -k4,4 -n | head -n 25 | awk -v dataset="$dataset" '{print "data_"dataset"/FILES_trimmed/"$2".fasta.mafft.clipkit"}' > best_scoring_${dataset}_${metric}.txt
else
echo "$metric_dat" | sort -k4,4 -nr | head -n 25 | awk -v dataset="$dataset" '{print "data_"dataset"/FILES_trimmed/"$2".fasta.mafft.clipkit"}' > best_scoring_${dataset}_${metric}.txt
fi
pk_create_concat -a best_scoring_${dataset}_${metric}.txt -p best_scoring_${dataset}_${metric}
done ;
done
2) What are the three files that the create_concat function generates? create_concat function in PhyKIT.
Hint
The answer is in the help message.
Solution
PhyKIT will output three files:
1) A fasta file with '.fa' appended to the prefix specified
with the -p/--prefix parameter.
2) A partition file ready for input into RAxML or IQ-tree.
3) An occupancy file that summarizes the taxon occupancy
per sequence.
3) Infer phylogenetic trees for each concatenation matrix generated using phylogenomic subsampling. To accelerate computation, use IQTREE with the -fast argument. Similarly, for the mammalian dataset, use the GTR+F+G4 substitution model and, for the yeast dataset, use the LG+F+G4 model. For reproducibility, use the seed 2016, which is specified with the -seed argument.
Note, this step may take time. It is another great opportunity to check in on your neigbor!
Solution
for dataset in mammals yeast ; do
for fasta in *${dataset}*fa ; do
[ "$dataset" == "mammals" ] && iqtree2 -s $fasta -pre $fasta -m GTR+F+G4 -fast -seed 2016 || iqtree2 -s $fasta -pre $fasta -m LG+F+G4 -fast -seed 2016
done
done
4) Determine if the phylogenetic trees inferred using phylogenomic subsampling differ from the species tree. To do so, calculate Robinson-Foulds metric, a measure of distance between two phylogenetic trees.
Hint
The paths for the species tree for mammals and yeast are ./data_mammals/Mammals_2002_eMRC.tree and ./data_yeast/Yeasts_2832_eMRC.tree, respectively.
Solution
for dataset in mammals yeast ; do
for tree in *${dataset}*treefile ; do
rf=$([ "$dataset" == "mammals" ] && pk_rf_dist ./data_mammals/Mammals_2002_eMRC.tree $tree || pk_rf_dist ./data_yeast/Yeasts_2832_eMRC.tree $tree) ;
echo -e "${dataset}\t${tree}\t${rf}" | sed -e 's/best_scoring_mammals_//g' -e 's/best_scoring_yeast_//g' -e 's/.fa.treefile//g' ;
done ;
done
mammals abs 6 0.1429
mammals aln_len 6 0.1429
mammals lbs 6 0.1429
mammals rcv 6 0.1429
mammals saturation 2 0.0476
mammals treeness 6 0.1429
mammals treeness_over_rcv 6 0.1429
yeast abs 0 0.0
yeast aln_len 0 0.0
yeast lbs 0 0.0
yeast rcv 0 0.0
yeast saturation 0 0.0
yeast treeness 0 0.0
yeast treeness_over_rcv 0 0.0
5) Based on these results, which phylogenetic tree is more unstable, the mammalian or yeast phylogeny?
Solution
The mammalian phylogeny is more unstable because some of the species trees inferred using phylogenomic subsampling differ compared to the species tree.
6) Determine what branch (or branches) in the mammalian phylogeny are unstable. To do so, use the -sup argument in IQTREE and your favorite tree visualization program.
Hint
To use the -sup argument, you will also have to use the -t argument.
iTOL is a user-friendly tree visualization software but there are many choices.
Solution
cat *mammals*treefile > mammals_subsampling_trees.tre
iqtree2 -t mammals_subsampling_trees.tre -sup data_mammals/Mammals_2002_eMRC.tree
Here, I used iTOL to quickly visualize the phylogeny and branch support.
In the phylogeny below, the ancestal bipartition of Loxodonta africana and Dasypus novemcinctus is unstable.
Similarly, so as the successive branches in the ancestor of Felis catus and Sus scrofa.
Instability at these branches has been observed in other studies - e.g., Murphy et al., Science (2001).
Note, the phylogeny (and subsequent phylogenies) are rooted on the clade containing Monodelphis_domestica, Macropus_eugenii, and Sacrophilus harrsii.
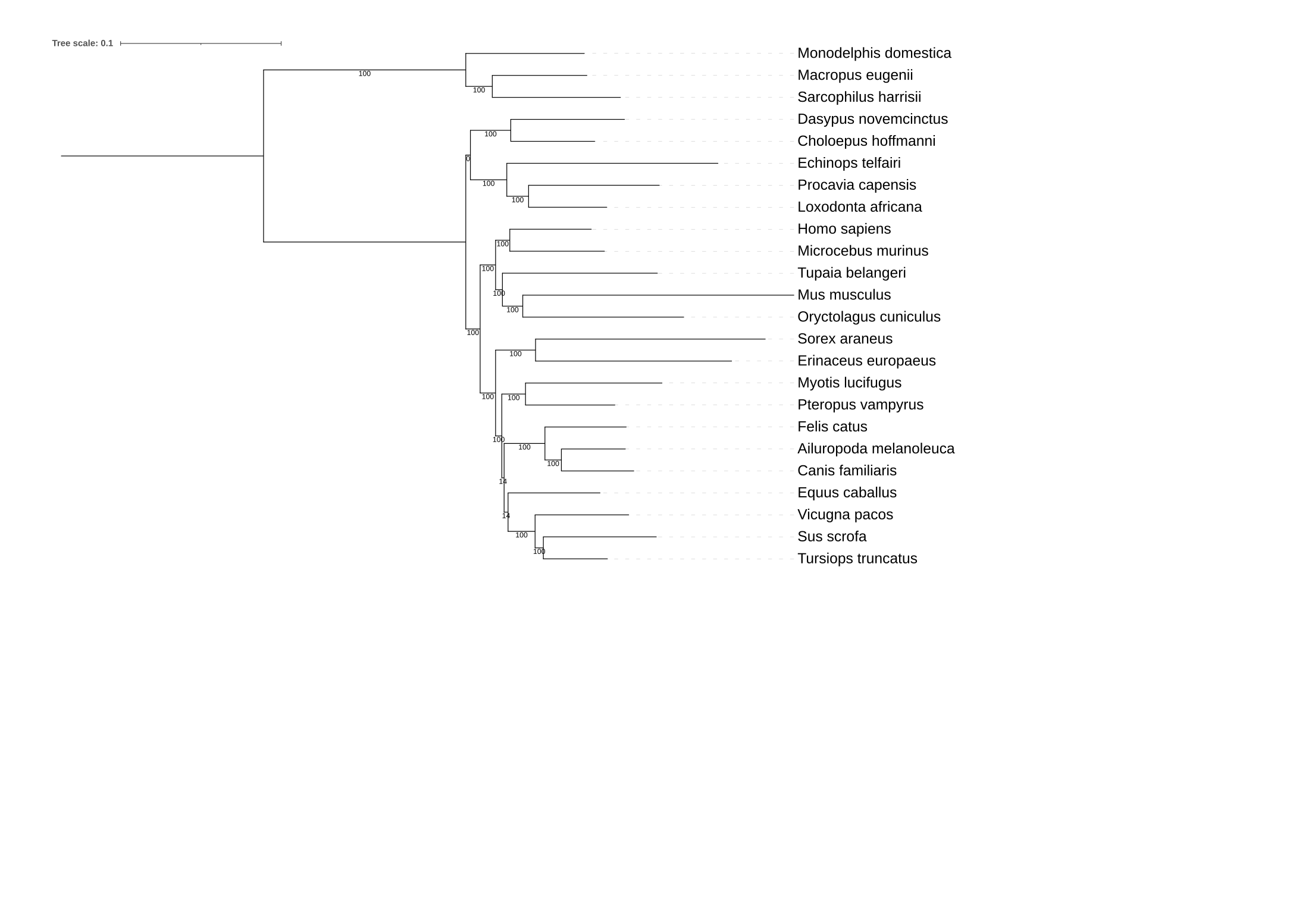
7) What are some reasons that could drive incongruence at these bipartitions?
Solution
1) Regarding the ancestal bipartition of Loxodonta africana and Dasypus novemcinctus, one reason for error may be that
many loci lack sufficient phylogenetic signal. Evidence supporting this idea stems from this branch being correctly recovered
among genes with the best average UFBoot scores.
2) Regarding, the ancestor of Felis catus and Sus scrofa, one reason for the lack of resolution may be successive
speciation events, which can manifest as numerous difficult-to-resolve sources of error. For example, successive short branches
are more susceptible to incomplete lineage sorting. Moreover, successive short branches may also be a signature of a radiation event.
Radiation events are well documented among mammals (e.g., Liu et al. PNAS (2017)).
3) Another reason may be that nucleotide sequences are insufficient at resolving these deeper bipartitions and that amino acids may be more appropriate.
4) *Note, in our analysis, we also only used 50 genes and subsampled 25. Thus, our dataset is very small compared to the typical hundreds of genes
that are common in phylogenomic analyses. Thus, some errors may also stem from insufficient gene sampling.
8) Bonus question: There is a common source of phylogenomic error not examined in these datasets. What do you think it is?
Solution
While there could be many answers to this question, I was specifically thinking of taxon occupancy. In these datasets, all orthologs have all taxa present.
However, orthologs that have many missing taxa can contribute noise due to low taxon occupancy.
Of course, more broadly, insufficient taxon sampling may also be a source of error.
If you had another source of phylogenomic error in mind, share it with someone sitting next to you or a teaching assistant and get their opinion :D
Together, these values help explore the various dimensions of information content observed in single genes.
Section 3: Phylogenomic subsampling across taxa
3.1. Summarize information content across taxaThe objective of this section is to identify taxa that may potentially contribute to error during species tree inference.
1) Other metrics capture information content in a taxon-specific manner. This can be useful for diagnosing is any specific taxa may be problematic. These metrics include:
2) For each ortholog, calculate both metrics using PhyKIT. Save the output to a file titled info_content_taxa.txt. The output should have five columns.
Hint
Links to PhyKIT documentation for each metric are provided in the text above.
Solution
for dataset in $(echo "mammals yeast");
do
for i in $(ls data_${dataset}/FILES_trimmed/);
do
aln=$(echo "$i")
tre=$(echo "${i}.treefile")
id=$(echo "$i" | sed 's/.fa.*$//g')
pk_rcvt data_${dataset}/FILES_trimmed/${aln} | awk -v OFS='\t' -v dataset="$dataset" -v id="$id" '{print dataset, id, "rcvt", $0}'
pk_lbs data_${dataset}/FILES_trees/${tre} -v | awk -v OFS='\t' -v dataset="$dataset" -v id="$id" '{print dataset, id, "lbs", $0}'
done
done |tee info_content_taxa.txt
3) For each dataset, what three taxa have the best long branch score and RCVT values?
Hint
Modify the previous solution and use the sort function in UNIX.
Also, lower values are better.
Solution
for metric in $(echo "lbs rcvt");
do
for i in $(echo "yeast mammals");
do
grep "$i" info_content_taxa.txt | awk '{print $4}' | sort | uniq | \
while read taxon ;
do
avg=$(grep "$taxon" info_content_taxa.txt | grep "$metric" | awk '{s+=$5} END {print s/NR}') ;
echo -e "${metric}\t${i}\t${taxon}\t${avg}" ;
done | sort -k4,4 -n | head -n 3;
done ;
echo ""
done
lbs yeast Saccharomyces_cerevisiae -27.4952
lbs yeast Saccharomyces_kudriavzevii -27.2058
lbs yeast Saccharomyces_uvarum -26.9693
lbs mammals Tursiops_truncatus -27.1216
lbs mammals Equus_caballus -27.0803
lbs mammals Microcebus_murinus -25.5538
rcvt yeast Saccharomyces_mikatae 0.010044
rcvt yeast Saccharomyces_cerevisiae 0.01026
rcvt yeast Saccharomyces_kudriavzevii 0.010434
rcvt mammals Pteropus_vampyrus 0.00542
rcvt mammals Tursiops_truncatus 0.005624
rcvt mammals Equus_caballus 0.00637
# EXPLANATION:
For the mammals dataset,
Tursiops_truncatus, Equus_caballus, and Microcebus_murinus have the lowest long branch scores.
For the yeast dataset,
Saccharomyces_cerevisiae, Saccharomyces_kudriavzevii, and Saccharomyces_uvarum have the lowest long branch scores.
For the mammals dataset,
Pteropus_vampyrus, Tursiops_truncatus, and Equus_caballus have the lowest signature of compositional bias.
For the yeast dataset,
Saccharomyces_mikatae, Saccharomyces_cerevisiae, and Saccharomyces_kudriavzevii have the lowest signature of compositional bias.
Species mentioned herein are unlikely contributing to error due to long branch attraction artifacts or compositional bias.
4) For each dataset, what three taxa have the worst long branch score and RCVT values?
Hint
Modify the previous solution.
Solution
for metric in $(echo "lbs rcvt");
do
for i in $(echo "yeast mammals");
do
grep "$i" info_content_taxa.txt | awk '{print $4}' | sort | uniq | \
while read taxon ;
do
avg=$(grep "$taxon" info_content_taxa.txt | grep "$metric" | awk '{s+=$5} END {print s/NR}') ;
echo -e "${metric}\t${i}\t${taxon}\t${avg}" ;
done | sort -k4,4 -nr | head -n 3;
done ;
echo ""
done
lbs yeast Kluyveromyces_blattae 22.8012
lbs yeast Saccharomyces_naganishii 8.70599
lbs yeast Kluyveromyces_phaffii 1.33263
lbs mammals Sarcophilus_harrisii 70.2817
lbs mammals Monodelphis_domestica 61.5831
lbs mammals Macropus_eugenii 58.0574
rcvt yeast Kluyveromyces_blattae 0.016976
rcvt yeast Saccharomyces_naganishii 0.016662
rcvt yeast Kluyveromyces_polysporus 0.014382
rcvt mammals Choloepus_hoffmanni 0.014174
rcvt mammals Microcebus_murinus 0.012482
rcvt mammals Macropus_eugenii 0.011872
# EXPLANATION:
For the mammals dataset,
Sarcophilus_harrisii, Monodelphis_domestica, and Macropus_eugenii have the highest long branch scores.
For the yeast dataset,
Kluyveromyces_blattae, Saccharomyces_naganishii, and Kluyveromyces_phaffii have the highest long branch scores.
For the mammals dataset,
Choloepus_hoffmanni, Microcebus_murinus, and Macropus_eugenii have the highest signature of compositional bias.
For the yeast dataset,
Kluyveromyces_blattae, Saccharomyces_naganishii, and Kluyveromyces_polysporus have the highest signature of compositional bias.
Species mentioned herein may contribute to error due to long branch attraction artifacts or compositional bias.
3.2. Visualize information content from taxa
The objective here is to learn how to make (near) publication-ready figures that summarize the distributions of the taxon-level information content in both data matrices.
1) Using R, Python, or any other method for plotting, plot the distribution of each metric. Note, the solution will be in the R programming language since many scientists are familiar with it and there is extensive, easily accessible documentation for plotting.
Hint
The R package ggplot2 can be very helpful for plotting. The facet_wrap() function will be very helpful for this data structure (i.e., long format).
Solution
# load R packages
library(ggplot2)
library(ggpubfigs)
library(cowplot)
# read in the data
dat<-read.table("info_content_taxa.txt")
# rename the columns
colnames(dat) <- c("dataset", "gene", "metric", "taxon", "value")
# split data by dataset
mdat<-dat[dat$dat=="mammals",]
ydat<-dat[dat$dat=="yeast",]
# use ggplot2 and ggpubfigs to make the figure
mplot<-ggplot(mdat, aes(taxon, value)) +
geom_violin(fill="darkgrey") + geom_boxplot(width=0.2, fill="white", alpha=0.7) +
facet_wrap(~metric, scales="free_y") + theme_simple() +
scale_fill_manual(values = friendly_pal("ito_seven")) +
facet_wrap(~metric, scales="free_y") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1))
yplot<-ggplot(ydat, aes(taxon, value)) +
geom_violin(fill="darkgrey") + geom_boxplot(width=0.2, fill="white", alpha=0.7) +
facet_wrap(~metric, scales="free_y") + theme_simple() +
scale_fill_manual(values = friendly_pal("ito_seven")) +
facet_wrap(~metric, scales="free_y") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1))
plot_grid(mplot, yplot, nrow=2, align="htv")
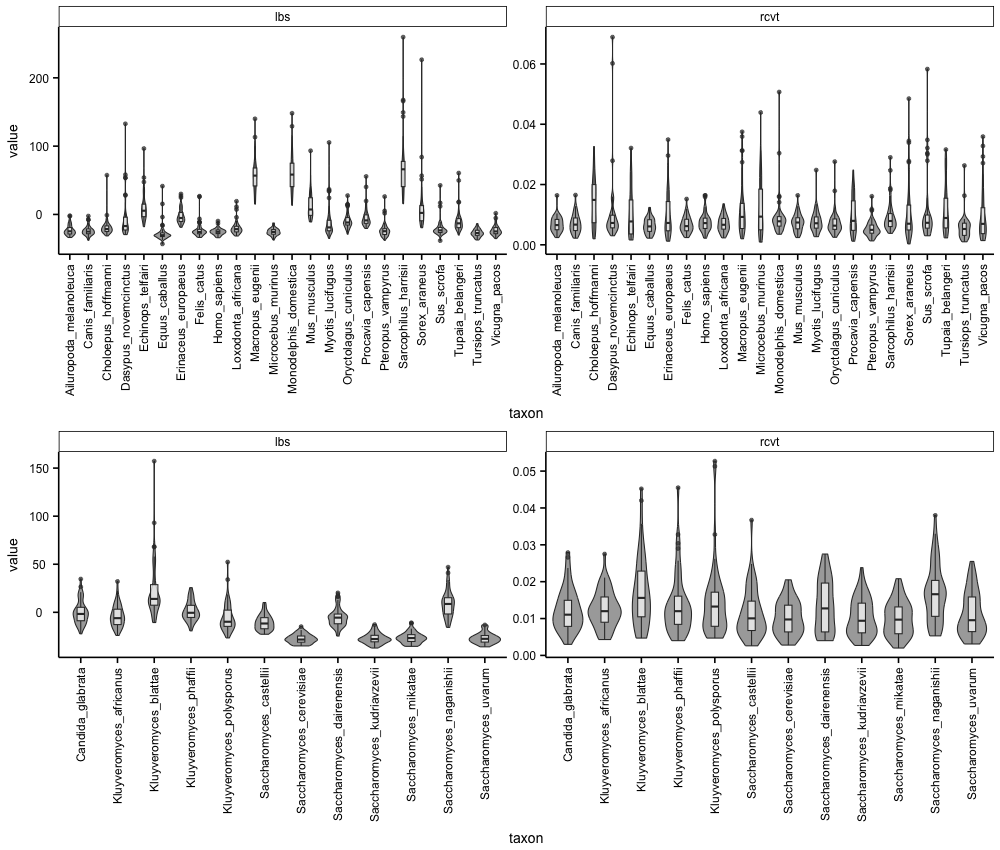
2) Examine the resulting distributions and determine if there are any other pontential outliers.
Solution
There are no other obvious outliers but visualization of the resulting distributions does indentify other taxa that may be problematic.
For example, in the mammalian dataset, Mus_musculus has a long branch score higher than some other taxa.
3.3. Create subsampled data matrices and infer phylogenies
Conduct phylogenomic subsampling analysis by removing the three taxa that have the worst long branch score or RCVT scores. Like the previous section, reinfer the species tree using a concatenation matrix that excludes that particular taxon.
Reinfer the species tree using IQTREE with the same models as in the previous section and with the -fast parameter.
Hint
Suggested workflow:
1) create a concatenation with all the data.
2) Remove individual taxa from that data matrix using the BioKIT function remove_fasta_entry
bk_remove_fasta_entry -e/--entry [-o/--output ]
3) Then, reinfer the species tree using IQTREE (set the seed to 2016 again)
To achieve this, one solution is to write one for loop for creating the concatenated matrices with all the data,
a second for loop to generate the matrices with the individual taxon pruned, and a third loop that infers a
phylogenetic tree from the resulting pruned matrices.
Solution
# generate concatenation matrices
for dataset in mammals yeast ; do
ls data_${dataset}/FILES_trimmed/* > ${dataset}_all_alns.txt ;
pk_create_concat -a ${dataset}_all_alns.txt -p ${dataset}_all_data ;
done
# generate pruned data matrix and infer a phylogenetic tree
for metric in lbs rcvt; do
for dataset in yeast mammals; do
grep "$dataset" info_content_taxa.txt | awk '{print $4}' | sort | uniq | \
while read taxon ; do
avg=$(grep "$taxon" info_content_taxa.txt | grep "$metric" | awk '{s+=$5} END {print s/NR}') ;
echo -e "${metric}\t${i}\t${taxon}\t${avg}" ;
done | sort -k4,4 -nr | head -n 3 | awk '{print $3}' | \
while read indiv ; do
echo "pruning $indiv from ${dataset}_all_data.fa only if ${dataset}_all_alns_${indiv}_pruned.fa does not exist"
[ ! -f "${dataset}_all_alns_${indiv}_pruned.fa" ] && bk_remove_fasta_entry ${dataset}_all_data.fa -e $indiv -o ${dataset}_all_alns_${indiv}_pruned.fa
done ;
done ;
done
# infer phylogenetic trees using the pruned datasets
for dataset in mammals yeast ; do
for fasta in *${dataset}*pruned.fa ; do
[ "$dataset" == "mammals" ] && iqtree2 -s $fasta -pre $fasta -m GTR+F+G4 -fast -seed 2016 || iqtree2 -s $fasta -pre $fasta -m LG+F+G4 -fast -seed 2016
done
done
3.4. Determine the impact of subsampling taxa on species tree inference
1) Using the rf_dist function in PhyKIT, determine if removing taxa had any impact on phylogenetic stability.
Solution
for dataset in mammals yeast ; do
for tree in *${dataset}*pruned.fa.treefile ; do
rf=$([ "$dataset" == "mammals" ] && pk_rf_dist ./data_mammals/Mammals_2002_eMRC.tree $tree || pk_rf_dist ./data_yeast/Yeasts_2832_eMRC.tree $tree) ;
echo -e "${dataset}\t${tree}\t${rf}" | sed -e 's/best_scoring_mammals_//g' -e 's/best_scoring_yeast_//g' -e 's/.fa.treefile//g' ;
done ;
done
mammals mammals_all_alns_Choloepus_hoffmanni_pruned 8 0.2
mammals mammals_all_alns_Macropus_eugenii_pruned 6 0.15
mammals mammals_all_alns_Microcebus_murinus_pruned 8 0.2
mammals mammals_all_alns_Monodelphis_domestica_pruned 6 0.15
mammals mammals_all_alns_Sarcophilus_harrisii_pruned 6 0.15
yeast yeast_all_alns_Kluyveromyces_blattae_pruned 0 0.0
yeast yeast_all_alns_Kluyveromyces_phaffii_pruned 0 0.0
yeast yeast_all_alns_Kluyveromyces_polysporus_pruned 0 0.0
yeast yeast_all_alns_Saccharomyces_naganishii_pruned 0 0.0
These observations indicate that, like the previous analysis, the mammalian dataset is sensitive to taxon sampling.
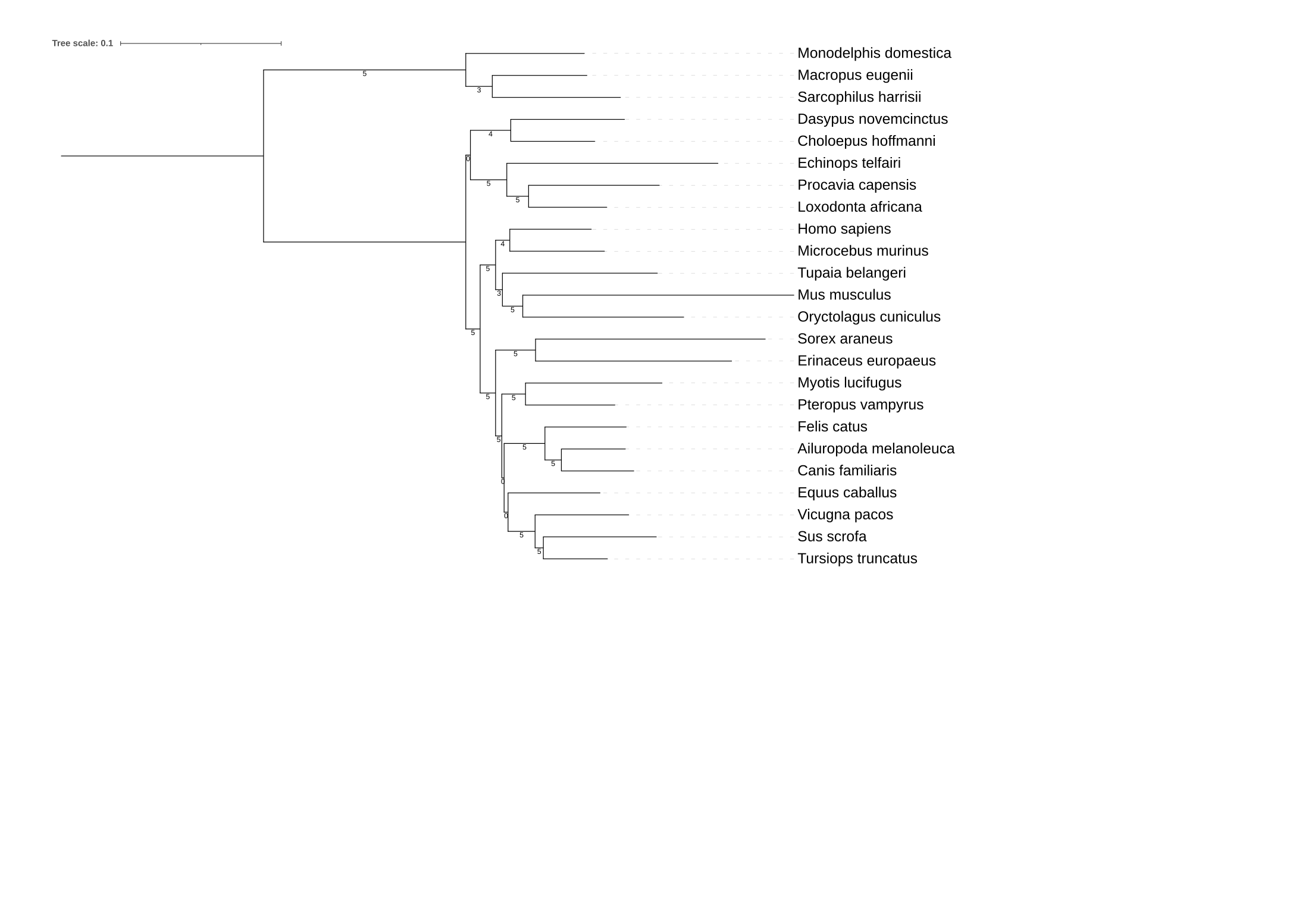
2) Determine what bipartitions are not well supported in the mammalian phylogeny. What is your interpretation of why these bipartitions are not well supported?
Hint
Like before, use the -sup argument in IQTREE to map single gene phylogenies back onto the species tree and
visualize the resulting phylogeny in your favorite tree viewing program.
Solution
# create a file with the phylogenies inferred using pruned taxa
cat mammals_*pruned*treefile > mammals_pruned_trees.tre
# map phylogenies inferred using pruned taxa onto the species tree
iqtree2 -t mammals_pruned_trees.tre -sup data_mammals/Mammals_2002_eMRC.tree
Visualize the resulting output, mammals_pruned_trees.tre.suptree, in your favorite
tree viewing software. Note that in this analysis bipartition support of 100% isn't possible
for all bipartitions due to taxon sampling.
After visualizing the phylogeny (which is also available if you scroll down), the unstable branches are largely the same as those identified in the gene subsampling analysis.
Thus, it is likely the similar drivers of incongruence are at play.
Lastly, it is important to underscore that the yeast phylogeny remains stable using this analysis. Therefore, researchers can
conclude that the yeast phylogeny is stable.
Section 4: Phylogenomic subsampling across sites
4.1. Summarize information content across sitesThe objective of this section is to determine if phylogenomic inference is sensitive to specific sites.
1) Numerous methods identify sites that may be sources of phylogenomic error. These metrics include:
2) For each concatenated matrix, calculate the evolutionary rate and compositional bias per site. Save all results to one file with the following structure:
Hint
Like before, use the -sup argument in IQTREE to map single gene phylogenies back onto the species tree and
visualize the resulting phylogeny in your favorite tree viewing program.
Solution
for method in comp_bias_per_site evo_rate_per_site ; do
for dataset in mammals yeast; do
pk_${method} ${dataset}_all_data.fa | awk -v OFS='\t' -v method="$method" -v dataset="$dataset" '{print dataset, method, $0}'
done ;
done |tee site_wise_info_content.txt
3) Next, we will remove between 1000 and 9000 sites (step of 4000) that are the fastest evolving or have the greatest degree of compositional bias. Typically, I would pick a much smaller step but we will use a large step to save time.
Additionally, there is no general consensus about how many sites should be removed during this analysis. The "best practice" will likely depend on many factors including the size of the full alignment. My personal recommendation would be to remove up to 20% of the full data matrix but, of course, as I just noted, this recommendation may vary depending on dataset size. To remove a list of sites from an alignment, use the ClipKIT cst trimming mode, which stands for custom trimming mode.
Hint
The cst method of trimming in ClipKIT requires a two column tab-delimited file. The first column is the site (starting with 1) and
the second column is if the site should be removed (specified with the string "trim") or kept (specified with the string "keep").
To streamline the user experience, researchers can also only specify which sites to trim (or keep) and ClipKIT will assume the others
should be kept (or trimmed).
For example, to trim the second position of an alignment with six sites, the tab-delimited file can be formatted in one of three ways:
format 1: specify what to do with every site
cat auxiliary_file.txt
1 keep
2 trim
3 keep
4 keep
5 keep
6 keep
format 2: specify what sites to trim
cat auxiliary_file.txt
2 trim
format 3: specify what sites to keep
cat auxiliary_file.txt
1 keep
3 keep
4 keep
5 keep
6 keep
Solution
for dataset in mammals yeast ; do
for method in comp_bias_per_site evo_rate_per_site ; do
for num_sites in `seq 1000 4000 9000` ; do
grep "$dataset" site_wise_info_content.txt | grep "$method" | sort -k4,4 -nr | head -n ${num_sites} | awk -v OFS='\t' '{print $3, "trim"}' > sites_to_trim_${dataset}_${method}_${num_sites}.txt ;
clipkit ${dataset}_all_data.fa -m cst -a sites_to_trim_${dataset}_${method}_${num_sites}.txt -o ${dataset}_${method}_${num_sites}sites_trimmed.fa ;
done ;
done ;
done
4.2. Infer phylogenies from subsampled data
Infer species trees from each trimmed dataset using IQTREE. As before, use the GTR+F+G4 model for the mammalian dataset and use the LG+F+G4 model for the yeast dataset. Similarly, use the -fast argument in IQTREE with the seed argument set to 2016.
Note, it is generally not best practice to use the same seed number over and over. We are only doing it for simplicity. Experimentally, it is best to assign a random number to the seed.
Hint
User previously written for loops to quickly infer phylogenetic trees
Solution
for dataset in mammals yeast ; do
for fa in ${dataset}*trimmed.fa ; do
[ "$dataset" == "mammals" ] && iqtree2 -s $fa -pre $fa -m GTR+F+G4 -fast -seed 2016 || iqtree2 -s $fa -pre $fa -m LG+F+G4 -fast -seed 2016 ;
done ;
done
4.3. Compare subsampled inferred trees to the species tree
1) Measure the Robinson-Foulds distance between each inferred phylogeny and the species tree.
Hint
Iterate on a written loop to achieve this aim.
Solution
for dataset in mammals yeast ; do
for method in comp_bias_per_site evo_rate_per_site ; do
for num_sites in `seq 1000 4000 9000` ; do
if [ "$dataset" == "mammals" ]; then
res=$(pk_rf_dist ${dataset}_${method}_${num_sites}sites_trimmed.fa.treefile ./data_mammals/Mammals_2002_eMRC.tree)
else
res=$(pk_rf_dist ${dataset}_${method}_${num_sites}sites_trimmed.fa.treefile ./data_yeast/Yeasts_2832_eMRC.tree)
fi
echo -e "${dataset}\t${method}\t${num_sites}\t${res}"
done ;
done ;
done
mammals comp_bias_per_site 1000 6 0.1429
mammals comp_bias_per_site 5000 6 0.1429
mammals comp_bias_per_site 9000 6 0.1429
mammals evo_rate_per_site 1000 4 0.0952
mammals evo_rate_per_site 5000 6 0.1429
mammals evo_rate_per_site 9000 4 0.0952
yeast comp_bias_per_site 1000 0 0.0
yeast comp_bias_per_site 5000 0 0.0
yeast comp_bias_per_site 9000 0 0.0
yeast evo_rate_per_site 1000 0 0.0
yeast evo_rate_per_site 5000 0 0.0
yeast evo_rate_per_site 9000 0 0.0
2) What is your interpretation of these results?
Solution
The mammalian phylogeny is sensitive to removing sites with compositional bias and fast evolutionary rates.
In contrast, the yeast phylogeny remains stable.
3) Map the resulting mammalian phylogenies onto the species tree using IQTREE. While mapping these results, make sure to map phylogenies from datasets pruned using each method separately. In other words, map phylogenies inferred using evolutionary rate as a subsampling method separately from phylogenies inferred using data subsampled based on compositional bias. Next, determine which bipartitions are unstable using your favorite visualization method.
Hint
As you have done before, use the -sup argument in IQTREE.
Also, among the many options you have, iTOL is a great user-friendly program to visualize phylogenetic trees.
Solution
for method in evo_rate_per_site comp_bias_per_site ; do
cat mammals_${method}*treefile > mammals_site_subsampling_${method}_trees.tre ;
iqtree2 -t mammals_site_subsampling_${method}_trees.tre -sup data_mammals/Mammals_2002_eMRC.tree ;
done
Below, the resulting phylogenies are depicted.
The first phylogeny is the tree with support values from phylogenies inferred using compositional bias
as a subsampling strategy. The second phylogeny has support values from phylogenies inferred from
mapping phylogenies inferred from subsampling the full data matrix based on site-wise evolutionary rate.
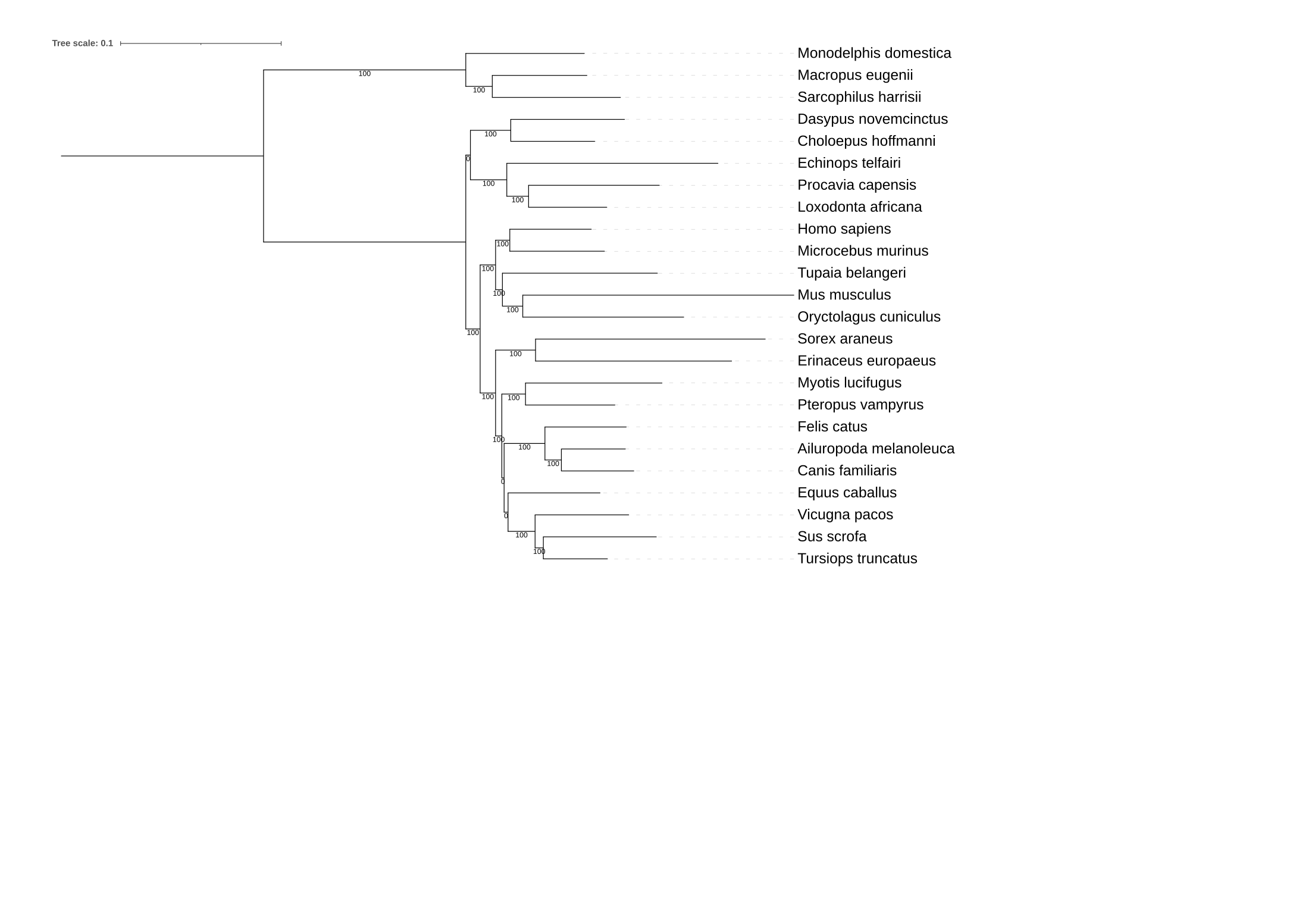
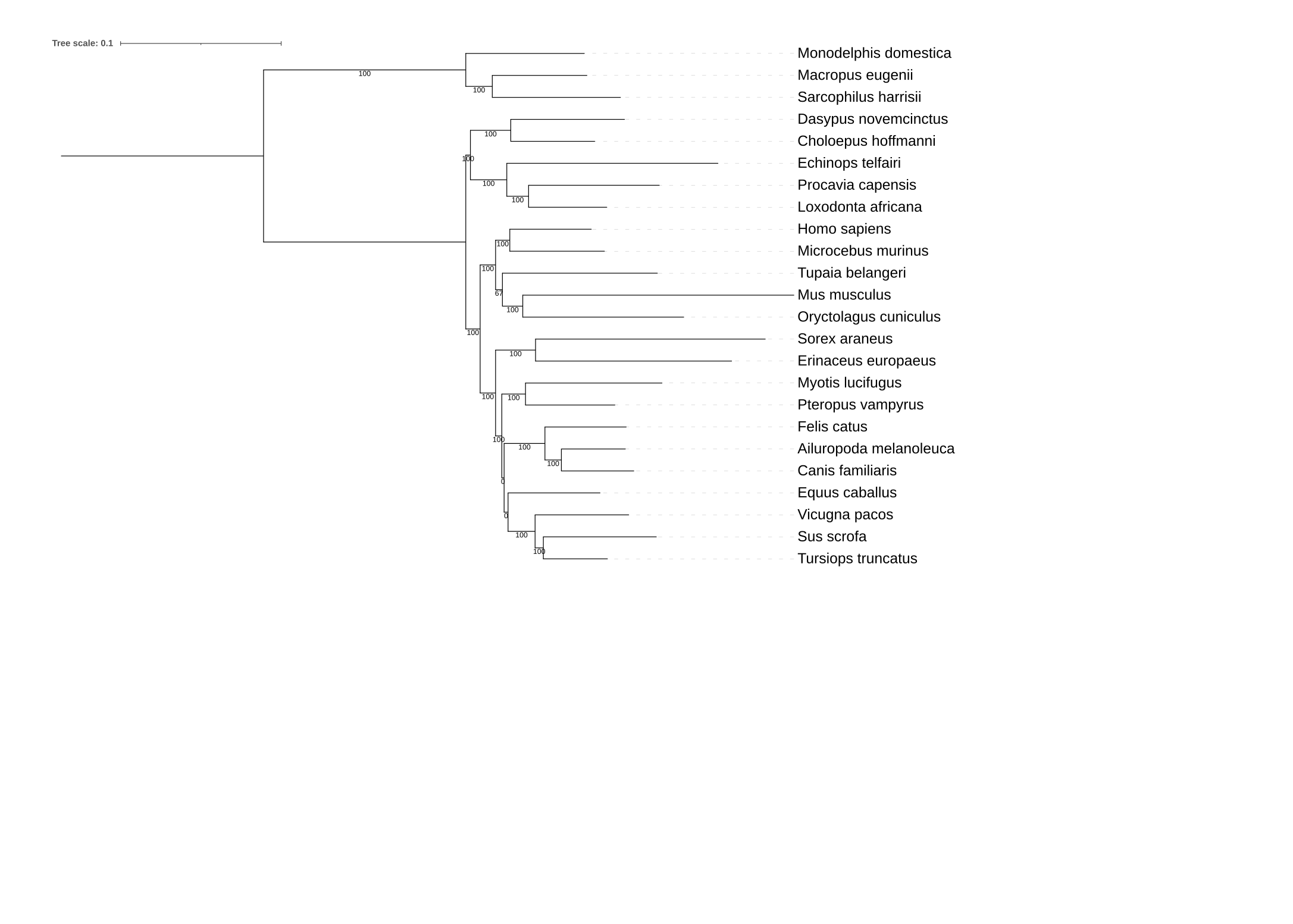
4) What is your interpretation of the results?
Solution
The same branches as identified before have signatures of incongruence. Thus, our interpretation remains the same.
However, in the analysis of the evolutionary rate-based subsampling, an additiona bipartition does not have full support.
This bipartition is at the ancestor of Tupaia belangeri and Musc musculus.
Closing remarks
Summary.
In this tutorial, we conducted phylogenomic subsampling at three levels: taxa, genes, and sites.
The workflow executed here can be used to identify unstable bipartitions in a phylogeny.
Unstable bipartitions warrant further dissection, which can lead to determining underlying drivers
of incongruence.
As a reminder, there are two classes for types of errors in phylogenomic studies:
Additional reading.
Ultimately, phylogenomic subsampling and sensitivity anlaysis is part of a large canon of work that aims to identify phylogenomically informative loci to help reconstruct the Tree of Life. Here is additional reading that follows a similar theme.
Acknowledgements.
This tutorial was written for the University of Barcelona advanced course titled Phylogenomics and Population Genomics: Inference and Applications. Lecture slides are available here. I especially want to thank the organizers for their invitation as well as Gemma I. Martínez-Redondo for her help in effectively teaching this tutorial. This includes providing feedback on earlier versions of this tutorial as well as helping students in real time. Thank you so much!
At the time of writing this tutorial, I am a Howard Hughes Medical Institute Awardee of the Life Sciences Research Foundation. Similarly, I am an advisor for Forensis Group Inc.
Thank you!
Thank you so much for taking this tutorial. Part of my goal is to democratize bioinformatics and I am excited by every opportunity to do so. So, sincerely, thank you for your time and attention throughout this section on phylogenomic subsampling.